Clear Sky Science · en
MaAsLin 3: refining and extending generalized multivariable linear models for meta-omic association discovery
Why tiny gut neighbors matter
Our bodies are home to trillions of microbes that help digest food, train the immune system and may even influence our mood. As DNA sequencing has made it easy to catalog these microbial communities, a crucial question has emerged: which specific microbes go hand in hand with diseases like inflammatory bowel disease, or with everyday traits such as age and diet? Answering this turns out to be surprisingly hard. The data are noisy, full of zeros and reported as percentages rather than true counts. This article introduces MaAsLin 3, a new statistical tool designed to pull clearer signals out of messy microbiome data so researchers can more reliably link microbes to human health and the environment.
Looking for patterns in a noisy crowd
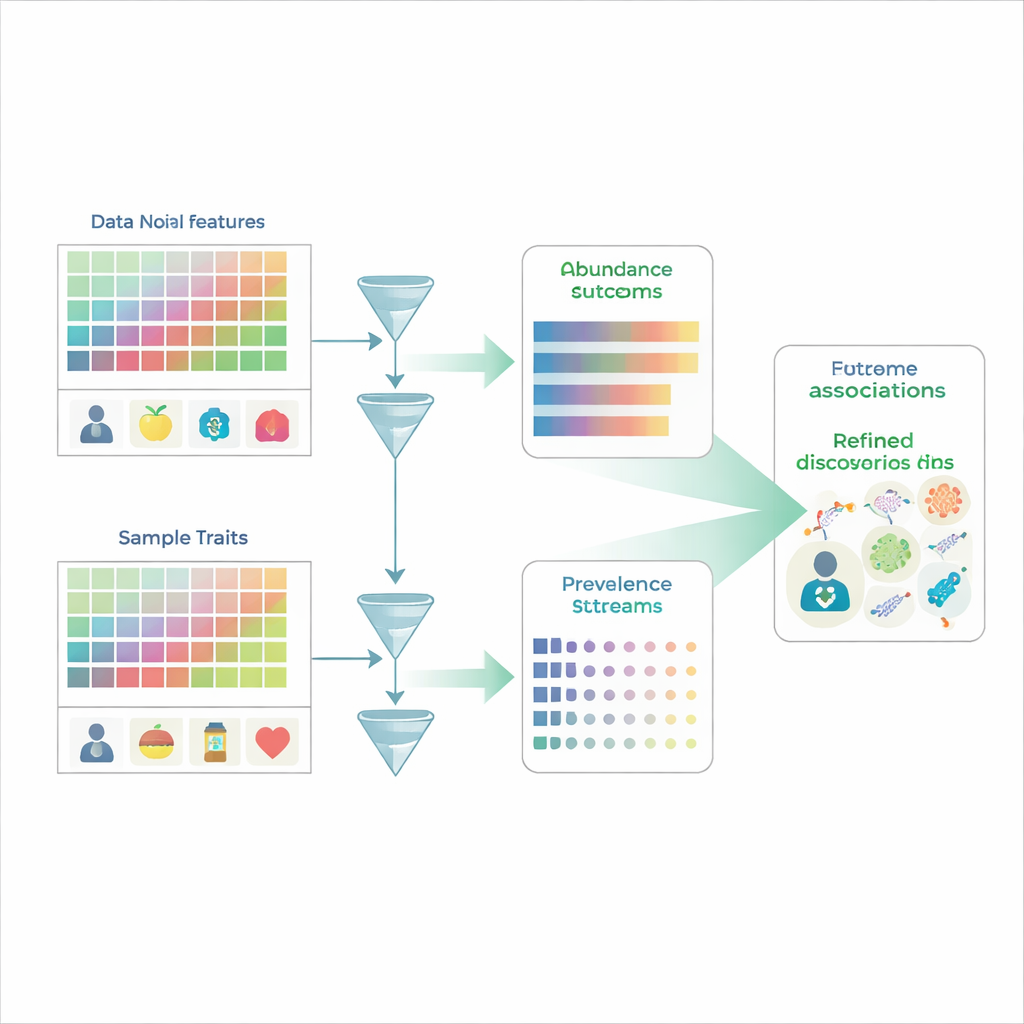
Traditional microbiome studies work a bit like counting faces in a crowd: researchers measure the relative abundance of hundreds or thousands of microbial species across many people, then ask which species differ between, say, sick and healthy groups. But microbiome data are constrained to percentages that must add up to 100%, so if one species goes up, at least one other appears to go down even if its true amount has not changed. On top of that, many species are simply not detected in a given sample, producing many zeros that may reflect either true absence or limits of detection. Common analysis methods typically blur together two distinct questions—whether a microbe is present at all, and how much of it is there when it is present—making it easy to misread the underlying biology.
Separating presence from amount
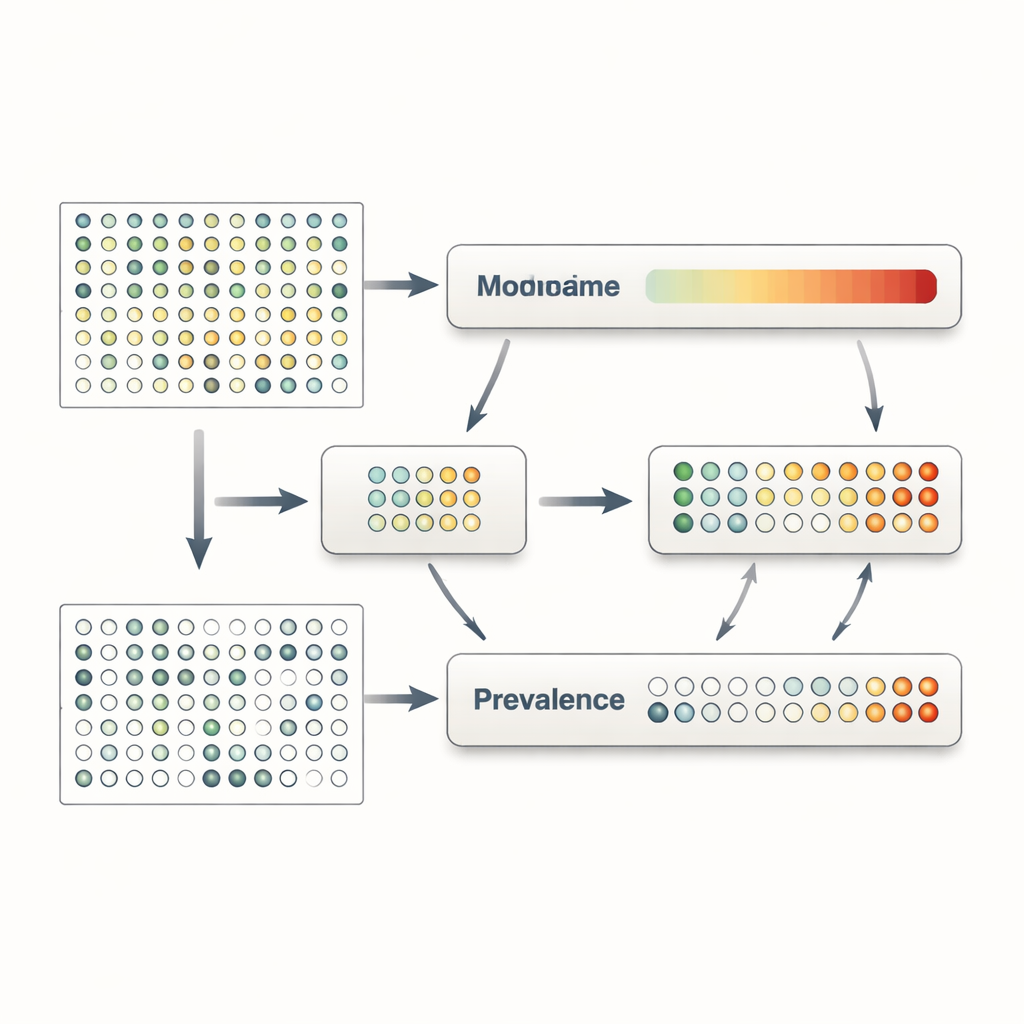
MaAsLin 3 tackles these problems by explicitly treating presence and amount as separate but related phenomena. For every microbial feature—such as a species, gene, or metabolic pathway—it builds two models in parallel. One model looks at prevalence, asking how often the feature is detected across samples with different traits. The other model focuses on abundance, asking how the feature’s level changes among only the samples where it is detected. By splitting the data this way, MaAsLin 3 avoids the common shortcut of filling in zeros with arbitrary small numbers, which can distort results. It then combines the two effects into an overall picture of how each feature relates to each trait, while still allowing researchers to see whether an association is mainly about presence, amount, or both.
Getting closer to real-world quantities
An additional complication in microbiome science is that most measurements are relative: they tell us what fraction of the total community a species occupies, not how many cells are actually there. Yet biological questions often depend on absolute abundance—for instance, whether a pathogen’s cell count crosses a threshold that might trigger disease. MaAsLin 3 offers two complementary solutions. When experiments include extra information, such as known quantities of a reference organism or estimates of total microbial load, the method can transform relative percentages into estimates of absolute counts and model those directly. When such data are not available, MaAsLin 3 instead compares each feature’s behavior to the typical pattern across all features, which under realistic assumptions approximates what would be seen on an absolute scale. Extensive computer simulations and tests on real datasets with experimentally measured absolute abundances show that this strategy accurately recovers underlying trends and outperforms several widely used tools.
Revealing hidden signals in gut disease
To show what these advances mean in practice, the authors applied MaAsLin 3 to a large, well-studied cohort of people with and without inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis. Earlier work had already identified many microbial changes in these conditions, but MaAsLin 3 added several layers of nuance. It confirmed most known links while clarifying that about three quarters of the associations involved changes in whether microbes were present at all, rather than in how abundant they were when present. In other words, gut inflammation often coincided with complete loss of certain helpful microbes or failure to detect them, rather than just a gentle decrease in their levels. The method also revealed microbes whose presence alone—regardless of how much of them was there—tracked strongly with disease-related disruption of the gut community.
What this means for future studies and care
For non-specialists, the key message is that the way we analyze microbiome data can dramatically shape which microbes we think matter for health. By better handling zeros, separating presence from amount and approximating real cell counts, MaAsLin 3 provides a sharper lens for discovering reliable microbial markers of disease, diet and environment. Its results in inflammatory bowel disease suggest that many clinically relevant shifts involve microbes that vanish or newly appear, not just those that slowly drift up or down in abundance. This distinction is important for designing therapies: if disease is tied to outright loss of beneficial species, strategies that reintroduce or protect those microbes may be more effective than approaches that simply try to nudge overall community balance. MaAsLin 3 thus equips researchers with a more precise and flexible toolkit for turning complex microbiome measurements into actionable biological insight.
Citation: Nickols, W.A., Kuntz, T., Shen, J. et al. MaAsLin 3: refining and extending generalized multivariable linear models for meta-omic association discovery. Nat Methods 23, 554–564 (2026). https://doi.org/10.1038/s41592-025-02923-9
Keywords: microbiome, inflammatory bowel disease, statistical modeling, absolute abundance, microbial prevalence