Clear Sky Science · en
CDK4/6 inhibition mitigates chemotherapy-induced expansion of TP53-mutant clonal hematopoiesis
Why protecting blood from cancer treatment matters
Cancer chemotherapy can be lifesaving, but it also batters the bone marrow where new blood cells are made. In some people, this damage accidentally encourages rare, pre‑existing mutant blood stem cells to take over, which can later lead to aggressive blood cancers. This study asks a hopeful question: can we use a temporary “pause button” drug on blood stem cells to shield them during chemotherapy, slowing the rise of these risky mutant clones without blunting cancer treatment itself?
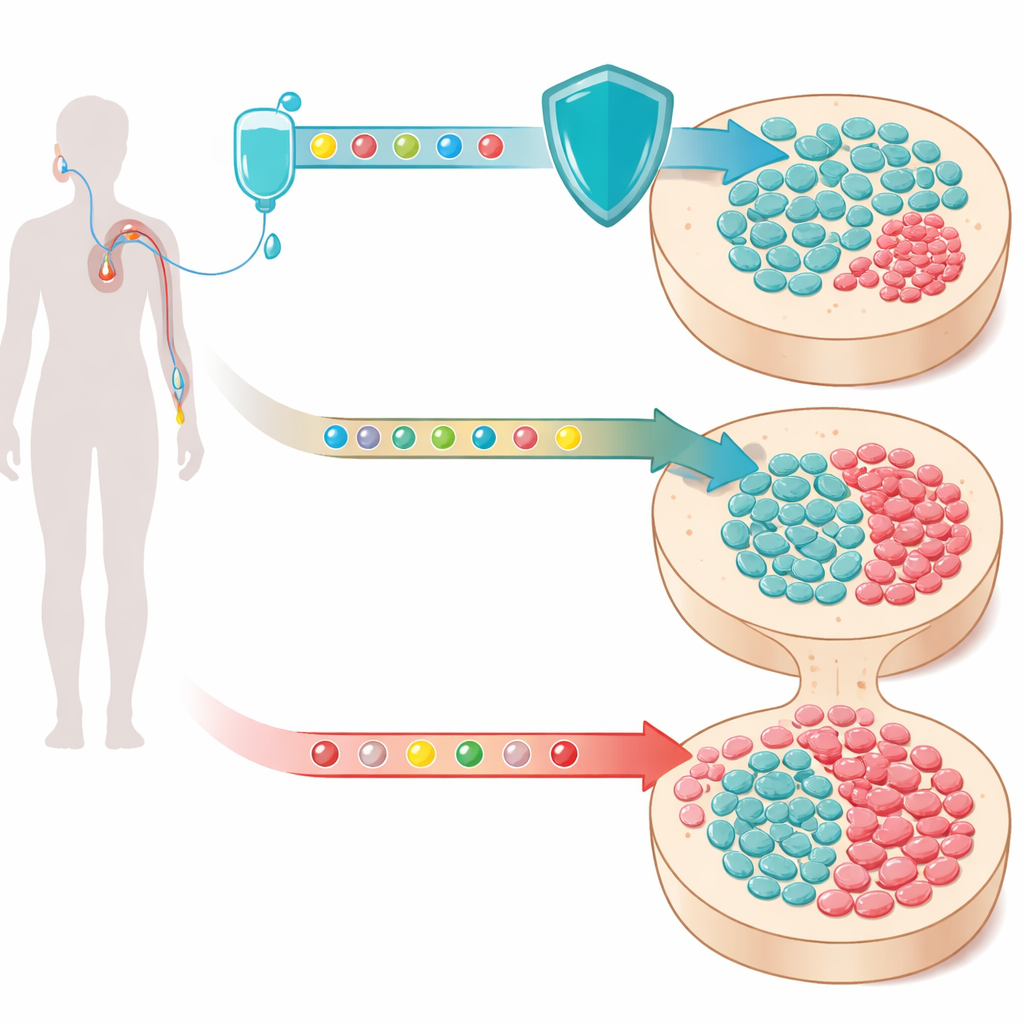
Hidden clones in the blood
As we age, our blood‑forming stem cells slowly collect DNA changes. Most of these altered cells stay harmless, but a few gain a growth edge and start forming small clones—pockets of genetically matched cells—circulating in the bloodstream. This phenomenon, called clonal hematopoiesis, is surprisingly common in older adults. When the affected gene is TP53 or other DNA damage‑response genes, those clones are especially worrisome: they survive stress that kills normal cells and are closely tied to therapy‑related myeloid neoplasms, a group of often fatal blood cancers that can appear years after chemotherapy.
Pressing pause on stem cells during chemotherapy
The researchers focused on drugs that block CDK4/6, key engines that drive cells to divide. One such drug, trilaciclib, is already approved to reduce low blood counts in people receiving certain lung‑cancer treatments. Given briefly before chemotherapy, it pushes bone‑marrow stem and progenitor cells into a resting state. The team reasoned that if both healthy and TP53‑mutant stem cells are less active when chemotherapy strikes, the mutants would lose much of their usual survival advantage, because fewer cells—normal or mutant—would be caught in the act of dividing when DNA‑damaging drugs are at their most toxic.
Evidence from cancer trials and animal models
To test this idea in real patients, the group analyzed blood samples from four randomized clinical trials in people receiving chemotherapy for small‑cell lung cancer, metastatic colorectal cancer, and triple‑negative breast cancer. In each trial, patients were randomly assigned to receive standard chemotherapy with either trilaciclib or placebo. Using ultra‑deep DNA sequencing of blood cells at the start of treatment and after several cycles, the scientists tracked how known mutant clones changed in size over time. Across all trials, clones carrying DNA damage‑response mutations—especially TP53 and PPM1D—expanded during chemotherapy, but they grew substantially more slowly in people who had received trilaciclib. On average, the growth rate of these risky clones was cut by roughly one‑third, while more routine age‑related mutations in other genes were largely unaffected.
Zooming in on how the protection works
Because patient follow‑up is still relatively short, the team turned to mouse models to uncover how CDK4/6 blockade reshapes the bone marrow during treatment. They created mice whose blood systems contained a small fraction of Trp53‑mutant stem cells, mimicking human clonal hematopoiesis. When these mice received platinum‑based chemotherapy alone, the mutant cells rapidly out‑competed their normal neighbors in both blood and marrow. But when trilaciclib—or a different CDK4/6 inhibitor, palbociclib—was given shortly before each chemotherapy dose, this mutant takeover was almost completely blocked. Detailed single‑cell RNA sequencing showed that CDK4/6 inhibition nudged stem and progenitor cells into a quieter, less proliferative state, reduced a “stemness” gene program that favors long‑lived mutant cells, steered development away from myeloid lineages and toward lymphoid ones, and selectively triggered cell death pathways in Trp53‑mutant stem cells while sparing normal ones.
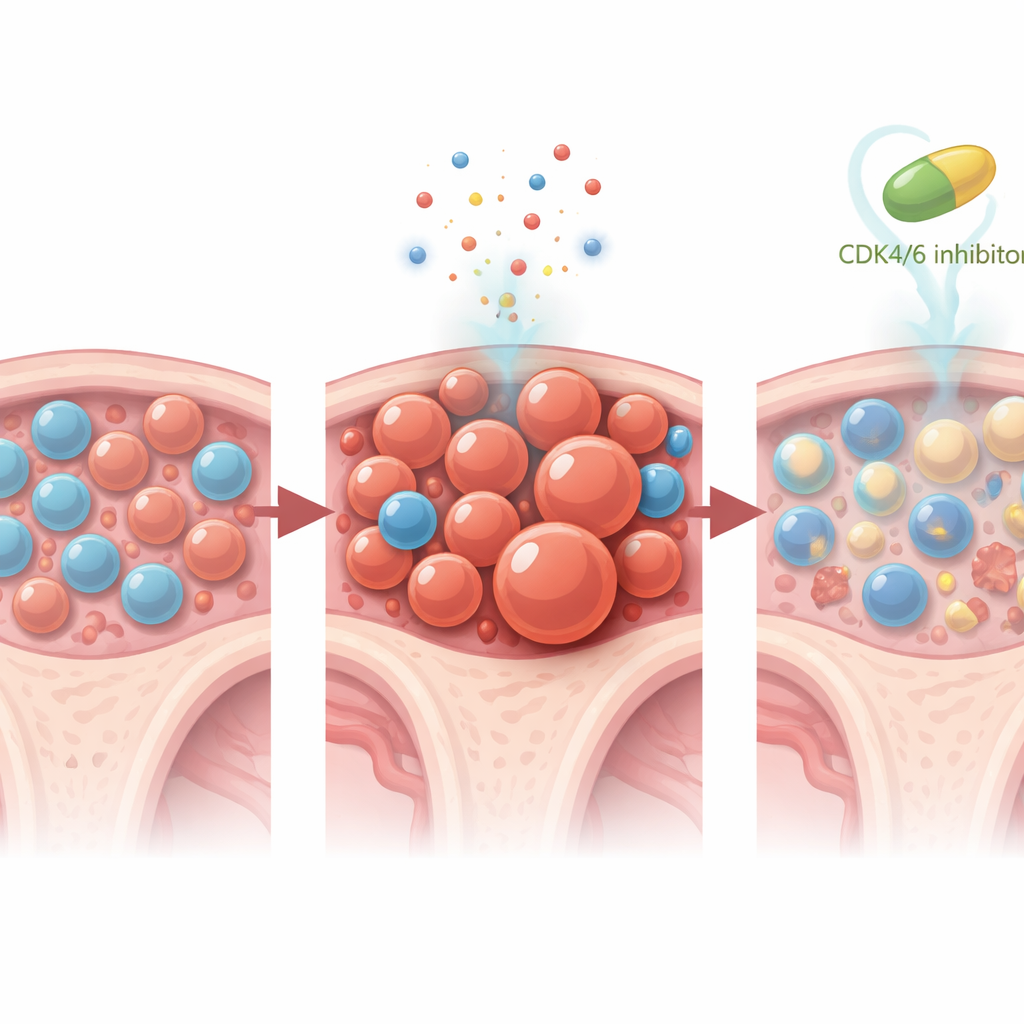
Lasting impact and future directions
One striking mouse experiment suggested that even a short course of CDK4/6 inhibition can have durable effects: two weeks of trilaciclib given around chemotherapy prevented the expansion of Trp53‑mutant clones for at least six weeks after all treatment had stopped. Importantly, blood counts and overall marrow health remained acceptable, indicating that the protective strategy did not simply trade one kind of toxicity for another. Although none of the patients in the trials developed blood cancer during the limited follow‑up, the presence and slowed growth of very small TP53‑mutant clones supports the idea that this is an early, modifiable step on the path to therapy‑related leukemia.
What this could mean for people with cancer
For patients who already harbor high‑risk blood cell clones, the fear has been that receiving the chemotherapy needed to control their solid tumor might also plant the seeds of a future, often untreatable leukemia. This work offers a proof of concept that carefully timed CDK4/6 inhibitors can blunt the growth advantage of those dangerous clones by briefly sheltering the bone marrow during chemotherapy. While longer and larger clinical studies are needed to prove that this strategy truly lowers the incidence of therapy‑related blood cancers, it points toward a future in which we can both treat the primary cancer aggressively and simultaneously protect the blood‑forming system from long‑term genetic harm.
Citation: Chan, I.C.C., Zhang, P., Pan, X. et al. CDK4/6 inhibition mitigates chemotherapy-induced expansion of TP53-mutant clonal hematopoiesis. Nat Genet 58, 582–592 (2026). https://doi.org/10.1038/s41588-026-02526-w
Keywords: clonal hematopoiesis, TP53 mutations, chemotherapy side effects, CDK4/6 inhibitors, therapy-related leukemia