Clear Sky Science · en
GGC repeat expansions within new open reading frames are translated into toxic polyglycine proteins in oculopharyngodistal myopathy
Hidden Messages in Our DNA
Most of us learned in school that only a small slice of our DNA actually codes for proteins, while the rest was once dismissed as “junk.” This study turns that idea on its head. It shows that small, overlooked stretches of repetitive DNA can secretly give rise to new proteins that damage muscles and the brain, helping to explain a group of rare but devastating neurological diseases—and pointing to a possible way to treat them.
Repetitive DNA and Mysterious Muscle Diseases
Our genome is full of tiny repeated sequences, like three-letter syllables copied over and over. When some of these repeats grow too long, they can cause more than 60 known human diseases. In oculopharyngodistal myopathy (OPDM) and a related disorder with brain changes called OPML, patients develop drooping eyelids, trouble swallowing, weakness in hands and feet, and sometimes broader nerve and brain problems. Under the microscope, doctors see distinctive clumps of protein inside muscle and nerve cells, but until now it was unclear how repeats sitting in supposedly “noncoding” regions of DNA could produce toxic proteins.
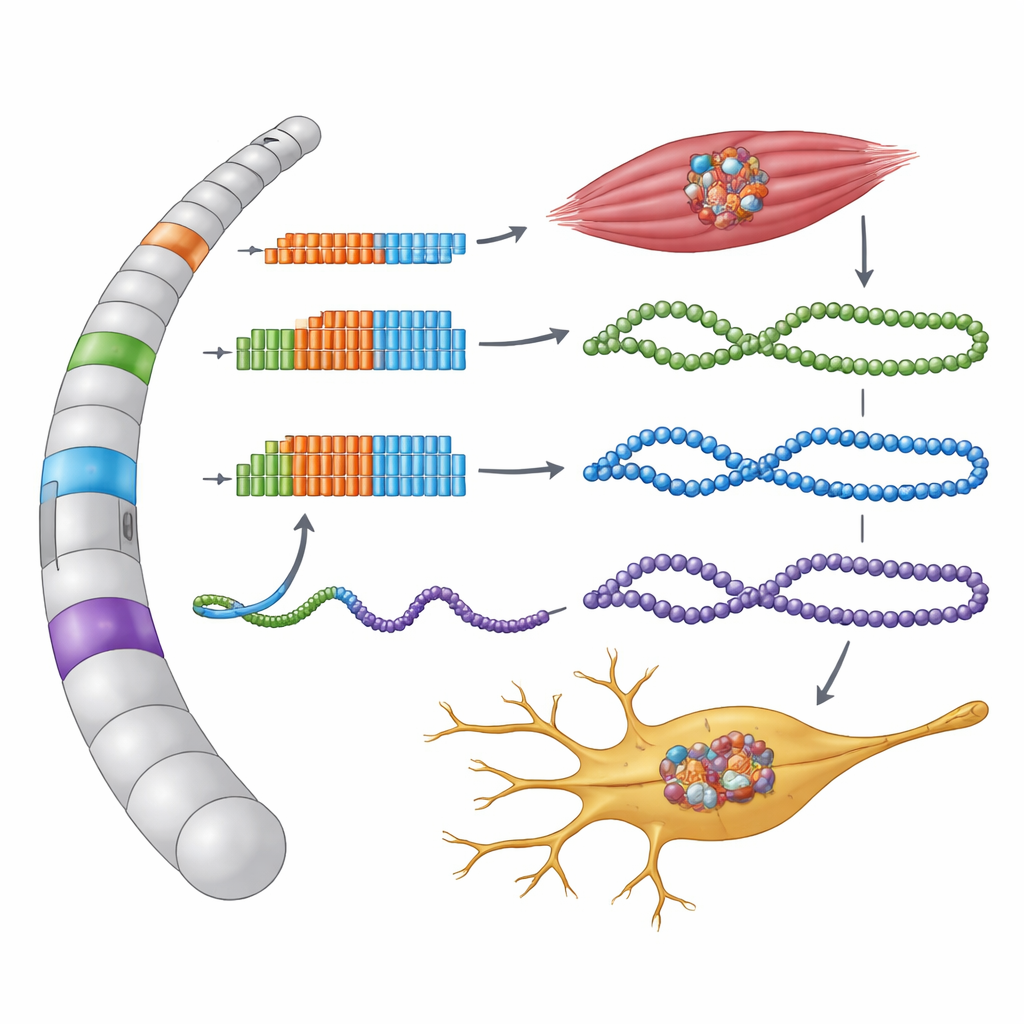
Noncoding Regions That Secretly Make Protein
The researchers focused on DNA regions where the three-letter sequence GGC is repeated many times within several genes linked to OPDM and OPML. These repeats lie in areas annotated as noncoding—untranslated tails of genes or long RNAs thought not to make protein at all. By recreating these human sequences in cells and tracking how they are read, the team discovered that each stretch of GGCs actually sits inside a tiny, previously unrecognized protein-coding unit called a small open reading frame. When cells read these hidden instructions, each GGC repeat is translated into the amino acid glycine, forming unusually long “polyglycine” tails on new microproteins.
New Toxic Proteins That Clump and Kill Cells
Using custom-made antibodies, the scientists showed that these polyglycine-bearing microproteins are present in muscle samples from patients and concentrate exactly where the strange p62-positive protein clumps are found. They then forced human muscle cells, flies, and mice to produce the same kinds of polyglycine proteins. In all three systems, the proteins condensed into round, dense inclusions in the cytoplasm and nucleus, resembling what is seen in patient tissues. Cells producing these proteins were more likely to die, and in mice the affected muscles showed shrinking fibers, internalized nuclei, and signs of inflammation. When the proteins accumulated in the brain and heart, animals developed movement problems, neurodegeneration, cardiomyopathy, and a shorter lifespan, matching many symptoms reported in patients.

One Core Toxic Feature, Many Local Flavors
Although these microproteins share the same central feature—a long chain of glycine residues—they are not identical. Each arises from a different tiny reading frame in a different gene and therefore has unique amino acid segments flanking the polyglycine stretch. The team found that these surrounding segments strongly influence how the proteins behave: where in the cell they accumulate, how readily they form aggregates, which cellular partners they interact with, and how toxic they are to muscle and nerve cells. Some variants were especially damaging, rapidly triggering inclusion formation and cell death, whereas others were somewhat milder. This suggests a common toxic core mechanism, fine-tuned by the local sequence context.
A First Step Toward a Shared Treatment Strategy
Encouragingly, the researchers also identified a small molecule, the cationic porphyrin TMPyP4, that can dial down both the buildup and the toxicity of these polyglycine proteins in cells and in a fruit fly model. TMPyP4 appears to act mainly by interfering with the translation of GC-rich repeat regions, reducing production of the harmful proteins without broadly shutting down protein synthesis. While far from a ready-made drug, it offers proof of principle that a single therapeutic approach might one day help patients with several related conditions driven by similar repeat expansions.
What This Means for Our Understanding of Disease
To a non-specialist, the central message is striking: stretches of DNA long written off as noncoding can hide tiny protein recipes that become dangerous when certain repeats expand. In OPDM, OPML, neuronal intranuclear inclusion disease and related disorders, those expanded GGC repeats are translated into sticky polyglycine proteins that clump inside cells and gradually impair muscles, nerves and brain. By uncovering this shared mechanism and a first candidate compound that can blunt it, the study broadens our view of what counts as a gene and opens new paths toward treating a growing family of repeat-driven neurological diseases.
Citation: Boivin, M., Yu, J., Eura, N. et al. GGC repeat expansions within new open reading frames are translated into toxic polyglycine proteins in oculopharyngodistal myopathy. Nat Genet 58, 517–529 (2026). https://doi.org/10.1038/s41588-026-02507-z
Keywords: oculopharyngodistal myopathy, microsatellite repeat expansion, polyglycine proteins, noncoding DNA translation, neurodegenerative muscle disease