Clear Sky Science · en
Transferable enantioselectivity models from sparse data
A Smarter Way to Find the Right Catalyst
Chemists often hunt for better medicines and materials by trying to stitch carbon atoms together in very specific three-dimensional arrangements. Getting that subtle "right-handed" versus "left-handed" outcome—known as enantioselectivity—usually means testing many metal catalysts and reaction conditions by trial and error. This paper introduces a way to use relatively small amounts of experimental data, combined with fast computer calculations, to predict which nickel-based catalysts will give the desired handedness in a wide range of reactions, potentially saving chemists weeks or months of lab work.
Why Handed Molecules Are So Hard to Control
Many drugs and natural products exist in mirror-image forms that can behave very differently in the body. Catalysts that favor one mirror image over the other are therefore extremely valuable. But designing such catalysts is tricky. Traditional quantum chemistry can, in principle, compute which pathway a reaction prefers, yet tiny energy errors translate into big mistakes in predicted selectivity, and the calculations are slow. Simpler statistical models, on the other hand, are fast but often ignore the detailed dance between the metal catalyst and the reacting molecules, especially when the reaction mechanism can subtly change as different partners are used.
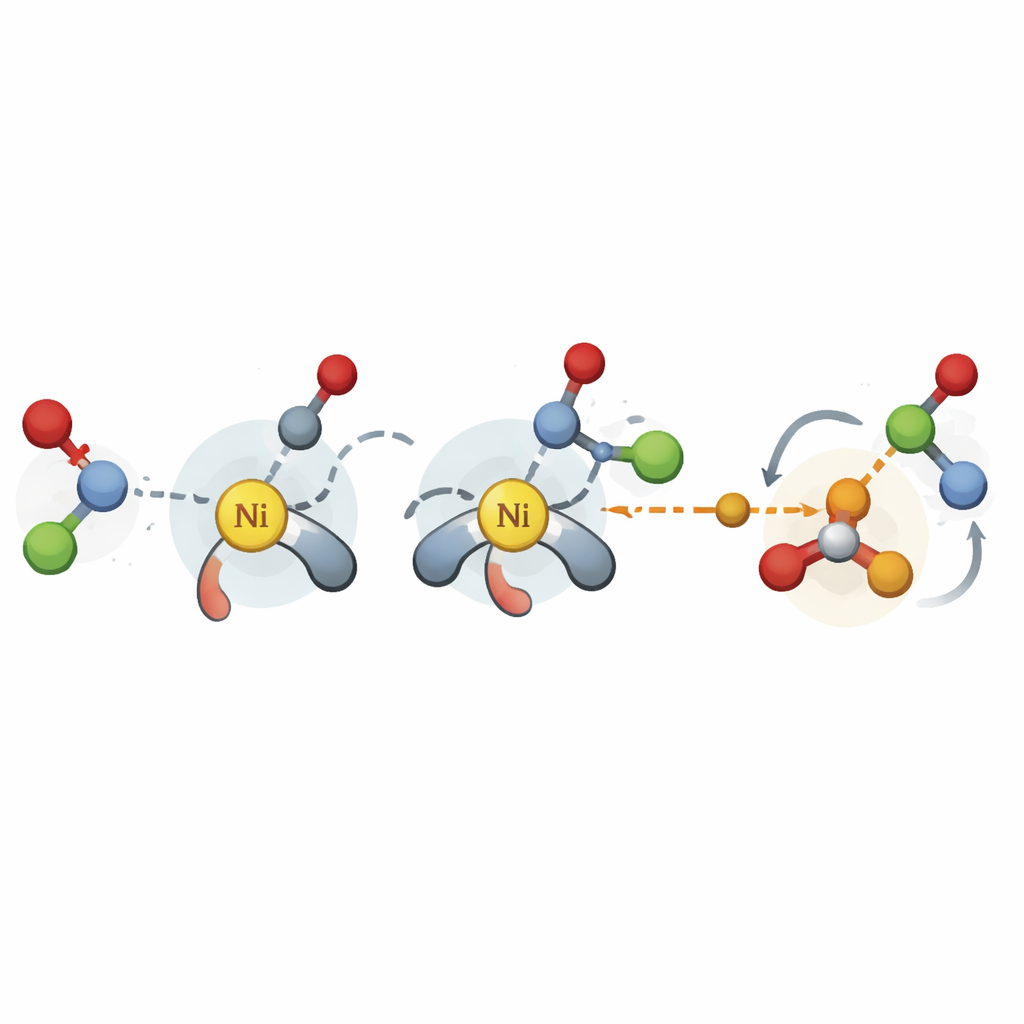
Capturing the Important Moments in a Reaction
The authors bridge this gap by focusing on the most critical stages of a nickel-catalyzed cross-coupling reaction: the steps where new carbon–carbon bonds are formed and the final product is released. Instead of running expensive high-level simulations, they use a streamlined quantum method to generate three-dimensional structures for key transition states and intermediates across many possible catalyst and substrate combinations. From these structures, they extract hundreds of physically meaningful descriptors, such as how crowded the catalyst environment is near certain atoms or how easily electrons can move. These numbers are then fed into straightforward linear regression models that connect structural features to the measured selectivity.
Learning from Sparse Data to Guide New Experiments
A central achievement of the work is that it makes the most of sparse data—the limited combinations of catalysts and substrates typically reported in a research paper. In one case study, the team revisits a nickel reaction that couples styrene oxides with aryl iodides. They show that descriptors taken from the most relevant transition state outperform those from simplified catalyst fragments, even though the underlying calculations are cheaper. With these models in hand, they virtually test many more ligands on existing substrate pairs and identify new catalyst choices that raise the enantiomeric excess for particularly stubborn examples, all while avoiding dozens of unnecessary experiments.
Transferring Knowledge Between Different Reactions
The approach is powerful because it can be transferred across different, yet related, nickel-catalyzed reactions. In a second set of studies, the authors combine data from several types of nickel reactions that all form bonds between sp3-hybridized carbon atoms and partners like aryl or alkenyl groups, even when the exact conditions or coupling partners differ. By building models from the same mechanistically meaningful descriptors, they successfully predict enantioselectivity for new ligands, new substrate combinations, and even an entirely new class of carbon–carbon bond-forming reaction that was not included in the training set. Analysis of which descriptors matter most also hints at which step in the catalytic cycle actually sets the handedness for each reaction family.
Helping Chemists Start New Reactions Faster
In a final demonstration, the authors use their descriptor scheme together with a Bayesian optimization platform to design a nickel-catalyzed coupling of benzylic acetals and aryl iodides that had not been developed asymmetrically before. Starting from literature data on other reactions, the model recommends small batches of promising ligands to test, quickly homing in on the best-performing class in only a few dozen experiments. For a chemist, this means a practical tool to "cold start" a new catalytic project: by feeding in a handful of early results, the model can suggest which chiral ligands are most likely to deliver high enantioselectivity. Overall, the study shows that thoughtfully chosen, low-cost computational features can turn limited past data into broadly useful guidance for building the next generation of selective reactions.
Citation: Gallarati, S., Bucci, E.M., Doyle, A.G. et al. Transferable enantioselectivity models from sparse data. Nature 651, 637–646 (2026). https://doi.org/10.1038/s41586-026-10239-7
Keywords: asymmetric catalysis, nickel cross-coupling, machine learning in chemistry, reaction optimization, enantioselectivity prediction