Clear Sky Science · en
Bidirectional CRISPR screens decode a GLIS3-dependent fibrotic cell circuit
When Healing Turns into Harmful Scarring
Our intestines are meant to repair themselves after every scrape and irritation. But in chronic diseases like Crohn’s disease and ulcerative colitis, this healing process can go off the rails, leading to thick, stiff scar tissue that narrows the gut and can require surgery. This study uncovers a hidden conversation between immune cells and structural cells in the intestine that drives this scarring, and pinpoints a master switch, a gene called GLIS3, that could offer a new way to break the cycle.
A Hidden Network Inside Inflamed Guts
To understand why some patients develop stubborn inflammation and fibrosis (scarring), the researchers created a cellular “atlas” of the human intestine. They combined single-cell RNA sequencing, which reads the genes active in individual cells, with spatial profiling that maps where those cells sit in real tissue slices. Using samples from people with Crohn’s disease, ulcerative colitis, and controls, they charted more than four million cells across the gut wall. Within this crowd, one fibroblast subgroup stood out: inflammation-associated fibroblasts, or IAFs. These cells congregated in areas of active and chronic colitis and carried a gene signature linked to resistance to standard anti–TNF therapies, suggesting they play a central role in hard-to-treat disease. 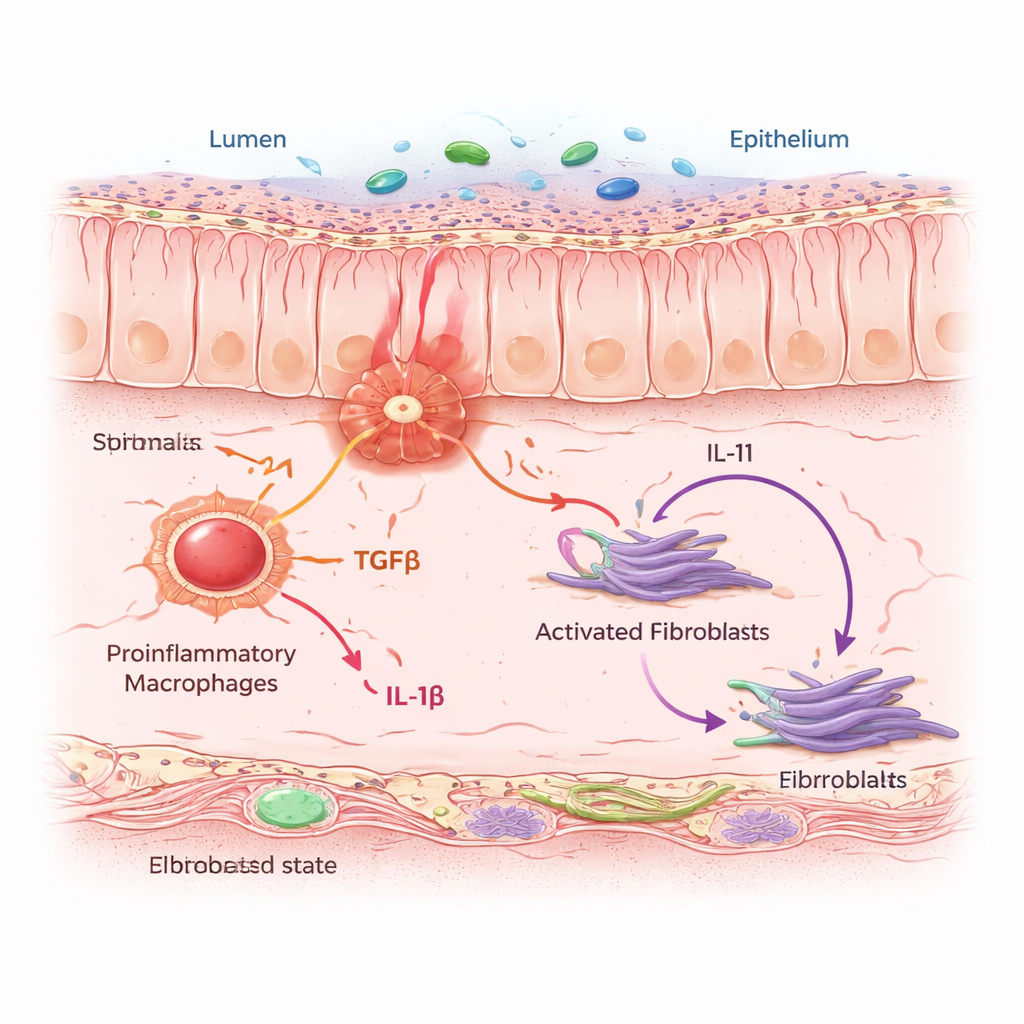
Macrophages Whisper, Fibroblasts Scar
IAFs did not act alone. They clustered in “neighborhoods” dense with proinflammatory macrophages—immune cells that sense danger and release alarm signals. Using computational models and cell co-culture experiments, the team showed that when macrophages are driven into an inflammatory state, they secrete two key messenger proteins: TGFβ and IL-1β. Nearby fibroblasts listen for these signals through specific receptors. When both signals arrive together, fibroblasts switch into the IAF state and start producing IL-11, a cytokine already suspected of promoting fibrosis, along with collagen and other matrix proteins that thicken and stiffen the intestinal wall. In mice exposed to a chronic colitis regimen, blocking IL-11 or deleting it selectively in fibroblasts reduced collagen buildup without preventing the initial inflammation, showing that IL-11 is a crucial driver of the scarring phase.
GLIS3: The Master Switch in Fibrotic Fibroblasts
To move from correlations to mechanisms, the authors used powerful genome-wide CRISPR tools. They engineered human fibroblasts so that IL-11 production could be monitored by a fluorescent tag, then performed parallel screens that either knocked genes out or turned them on across the genome. By sorting cells that made unusually high or low amounts of IL-11 after TGFβ and IL-1β stimulation, they identified genes that control this response. Among many signaling components, one transcription factor—GLIS3—emerged as a top regulator. When GLIS3 was disabled, fibroblasts produced far less IL-11; when it was boosted, IL-11 surged. Additional experiments showed that GLIS3 moves into the fibroblast nucleus in response to macrophage signals, binds directly to DNA near the IL11 gene and others, and activates a broad program of inflammatory and fibrotic genes, including collagens and factors that attract more immune cells. 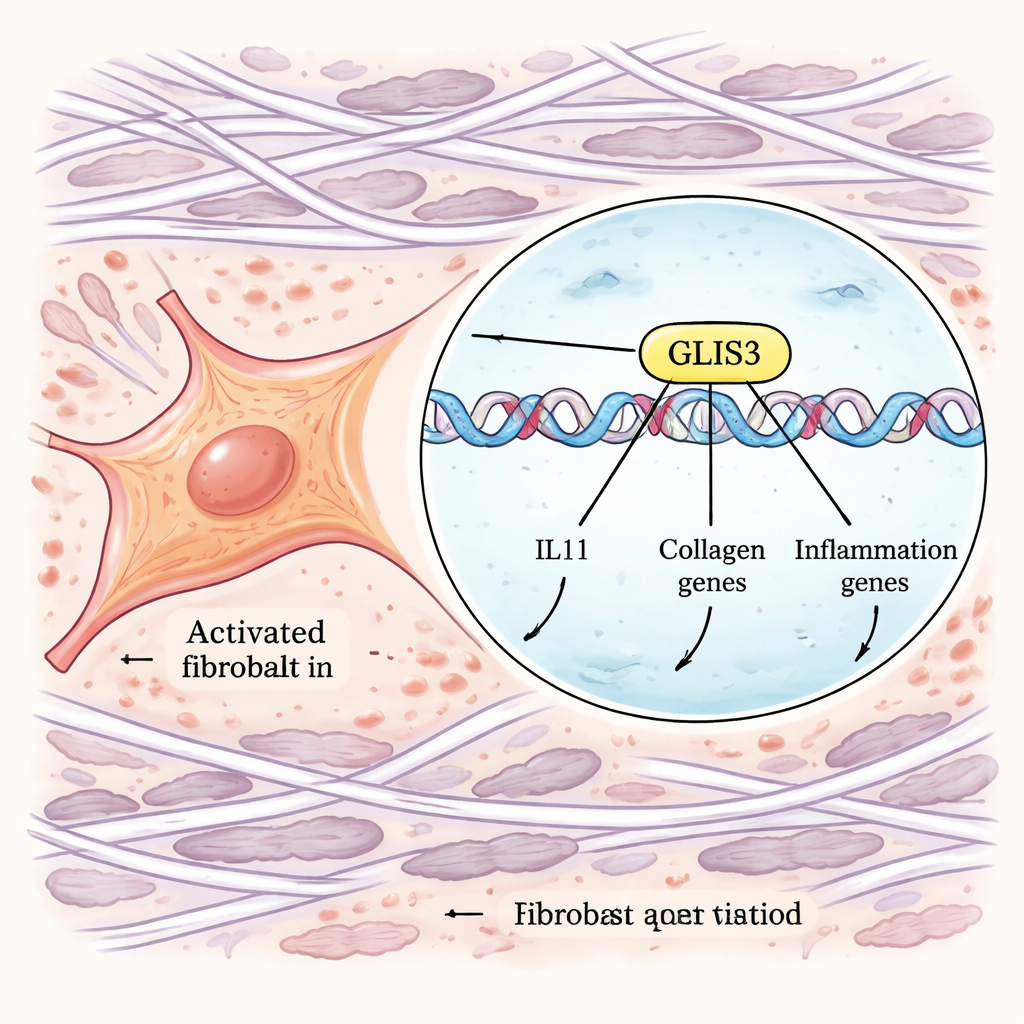
From Mouse Models to Patient Severity
The team then asked whether this GLIS3-driven program matters in living organisms. In mice, they created a strain in which GLIS3 could be removed only from fibroblasts. When these animals were subjected to chronic colitis, they developed less intestinal scarring, had lower levels of collagen and fibrotic gene expression, and showed reduced inflammation compared with normal mice. Spatial mapping confirmed that GLIS3-deficient mice had fewer IL-11–producing fibroblasts and fewer nearby activated macrophages and neutrophils, indicating that disrupting GLIS3 weakens the entire inflammatory-fibrotic circuit. Turning to a large pediatric ulcerative colitis cohort, the authors distilled a GLIS3 “signature” of 50 genes and found that its activity in colon biopsies closely tracked disease severity and the abundance of IAFs and activated macrophages, linking this pathway directly to patient outcomes.
Breaking the Cycle of Inflammation and Scarring
For non-specialists, the bottom line is that this work reveals a self-reinforcing loop: inflammatory macrophages trigger fibroblasts to become scar-forming IAFs; these IAFs, under the control of GLIS3, pump out IL-11, collagen, and other factors that remodel the tissue and attract more inflammatory cells. Standard drugs that broadly suppress the immune system may not fully disrupt this loop, which helps explain why many patients eventually fail existing therapies. By identifying GLIS3 and the IL-11–producing fibroblast state as central nodes in the inflammation–fibrosis circuit, this study points toward more targeted strategies—aimed at fibroblasts rather than only immune cells—that could one day prevent or reverse scarring in inflammatory bowel disease and possibly other chronic inflammatory conditions.
Citation: Pokatayev, V., Jaiswal, A., Shih, A.R. et al. Bidirectional CRISPR screens decode a GLIS3-dependent fibrotic cell circuit. Nature 650, 997–1006 (2026). https://doi.org/10.1038/s41586-025-09907-x
Keywords: inflammatory bowel disease, intestinal fibrosis, fibroblasts, macrophages, GLIS3