Clear Sky Science · en
Stress controls heterochromatin inheritance via histone H3 ubiquitylation
How Cells Remember Stress
Our cells need ways to "remember" past stresses—such as heat, lack of nutrients or drug exposure—so they can respond faster the next time. One powerful memory system relies on heterochromatin, tightly packed DNA that keeps certain genes turned off for many cell divisions. This paper explores how cells actively tune that memory in response to stress, revealing a molecular hub that links environmental signals to long‑lasting changes in gene activity. Understanding this control system could help explain how fungi evolve drug resistance and how stress reshapes our own cells’ epigenetic landscape.
A Lock on the Genome
Heterochromatin can be thought of as a molecular lock on stretches of DNA, keeping nearby genes silent. That lock is built from chemical tags on histone proteins, especially a mark called H3K9me3. Once established, it can copy itself every time DNA is replicated, allowing patterns of gene repression to be inherited without altering the underlying DNA sequence. Until now, most work suggested this self‑copying relies mainly on a "read–write" feedback loop: an enzyme complex recognizes existing H3K9me3 marks and adds the same mark to neighboring histones, gradually spreading the silent domain. But this model could not fully explain how environmental conditions speed up, slow down or redirect the spread of heterochromatin.
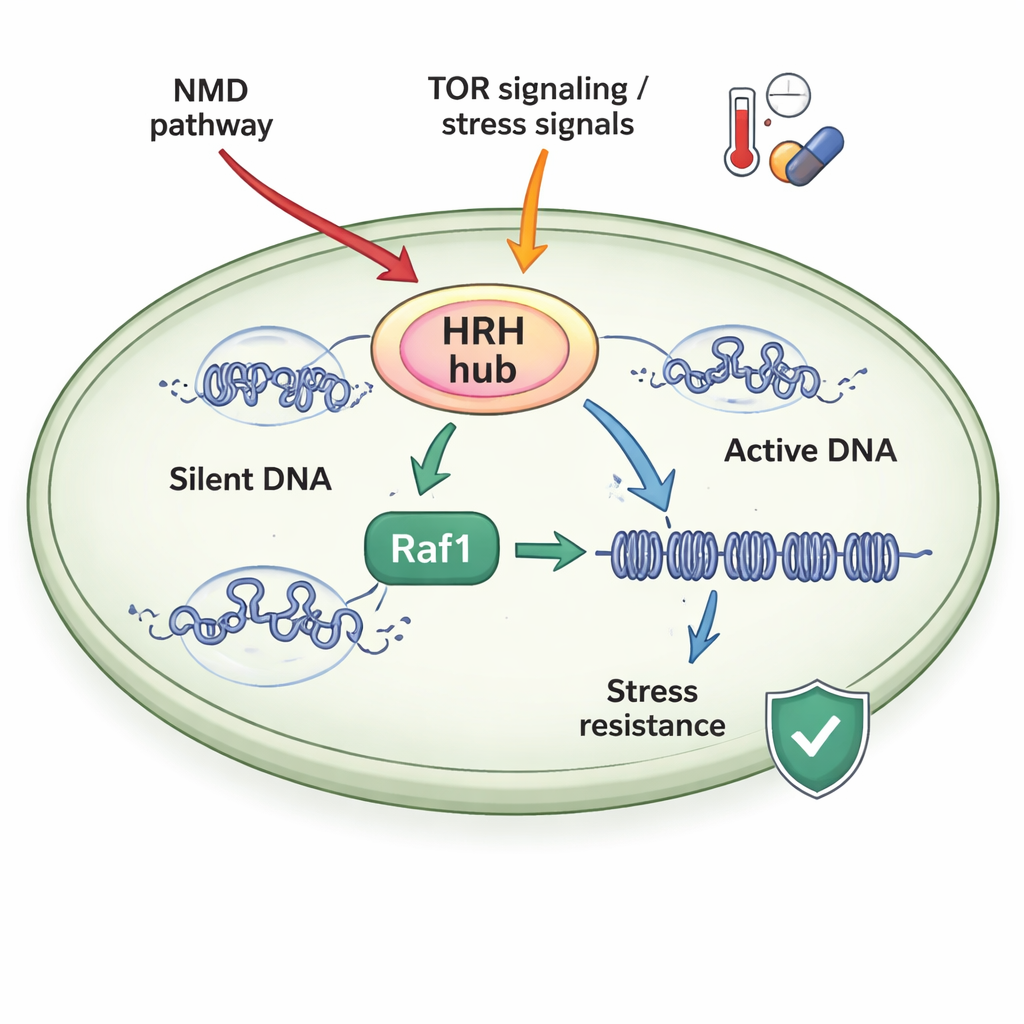
A Stress‑Sensitive Control Hub
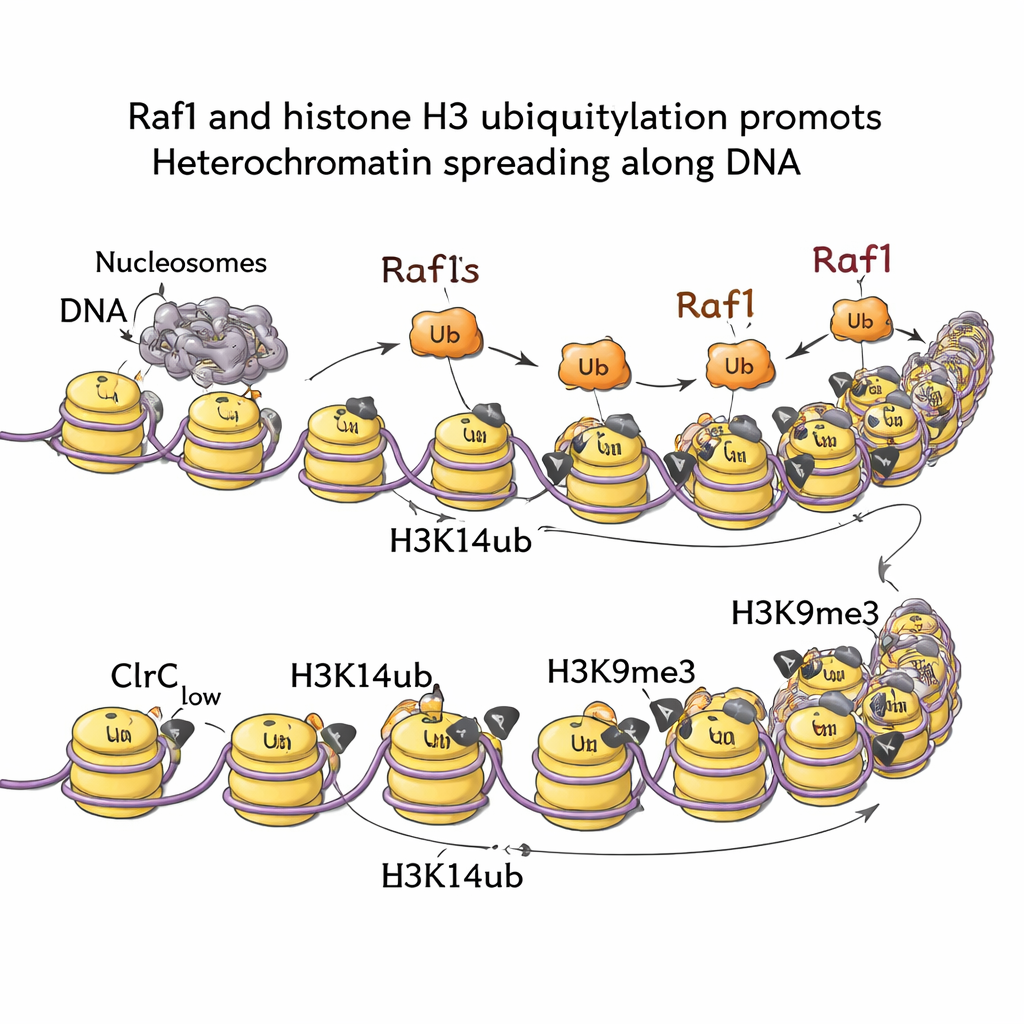
The authors studied fission yeast, a simple model organism whose chromatin machinery closely resembles that of higher organisms. They focused on a complex called ClrC, which both writes the H3K9me3 mark and attaches a small protein, ubiquitin, to another position on histone H3 (H3K14ub). A key subunit of ClrC, Raf1, acts as a limiting “gatekeeper”: when Raf1 is scarce, much of the main enzyme (Clr4) floats free instead of joining the complex on chromatin, and silent domains fail to spread. When Raf1 is abundant, more ClrC is assembled and stably bound to DNA, allowing H3K14ub and H3K9me3 marks to extend along chromosomes and reinforce gene silencing.
Fine‑Tuning the Lock With Ubiquitin
By mapping chromatin marks across the genome, the team showed that H3K14ub is highly enriched wherever heterochromatin forms and that this mark essentially disappears if Raf1 is removed. When cells carry a mutation that blocks the ubiquitin‑adding step (while leaving H3K9me3 intact at the starting point), silent domains are unable to spread outward. In other words, H3K14ub is not just decorative; it is required to push the heterochromatin front forward. Biochemical and imaging experiments reveal why: H3K14ub greatly boosts the activity of the Clr4 enzyme and helps keep the whole complex parked on chromatin, raising the local density of H3K9me3 above the threshold needed for stable inheritance. Remarkably, increasing Raf1 levels can bypass several other factors normally needed to maintain these silent domains, underscoring that Raf1‑driven ubiquitylation is a central control lever.
Signals From RNA Decay and Growth Pathways
Cells do not leave Raf1 levels to chance. The study shows that two major stress‑responsive systems feed into what the authors term a heterochromatin heritability regulatory hub (HRH), centered on Raf1. First, the nonsense‑mediated decay (NMD) pathway—best known for destroying faulty messenger RNAs—targets the messenger encoding Raf1, keeping Raf1 levels low in normal conditions. Disabling NMD stabilizes Raf1 RNA, raises Raf1 protein abundance and restores heterochromatin spreading in mutants that otherwise cannot maintain silent domains. Second, a growth and stress sensor called TORC2, acting through the kinase Gad8, promotes Raf1 expression. High temperature turns off this pathway, lowering Raf1 levels, weakening heterochromatin and making it harder for cells to maintain silent states unless Raf1 is experimentally boosted.
Stress, Drug Resistance and Broader Implications
The authors then connect this molecular hub to real‑world adaptation. Exposing yeast to caffeine, a stress that is also known to blunt NMD in other systems, raises Raf1 levels and enhances the spread of heterochromatin to new sites, including genes whose silencing confers resistance to caffeine and antifungal drugs. Likewise, cells with artificially elevated Raf1 become more resistant to fluconazole and clotrimazole, common antifungal agents. Conversely, when Raf1 is reduced—by heat or by loss of TORC2–Gad8 signaling—heterochromatin becomes unstable and epigenetic memory fades, unless Raf1 is restored. Because Raf1‑like proteins, the ClrC complex, and the H3K14ub mark all have counterparts in pathogenic fungi and in mammals, these findings suggest that a similar stress‑sensing epigenetic hub may shape drug resistance, development and disease in many species.
Why This Matters
Seen in everyday terms, this work shows that gene silencing by chromatin is not a rigid lock but a smart, adjustable system. Cells use a central hub to read environmental cues—temperature shifts, nutrient status, chemical stresses—and dial Raf1 up or down. That, in turn, governs how much of the genome is wrapped in long‑lasting silence and how easily cells can "reprogram" themselves without mutating their DNA. By uncovering the key role of histone H3 ubiquitylation and Raf1 dosage in this process, the study provides a blueprint for how stress can rapidly reshape the epigenetic landscape—and hints at new ways to influence drug resistance in fungi or aberrant gene silencing in human disease.
Citation: Bhatt, B., Wei, Y., Pradhan, A.K. et al. Stress controls heterochromatin inheritance via histone H3 ubiquitylation. Nature 650, 768–778 (2026). https://doi.org/10.1038/s41586-025-09899-8
Keywords: heterochromatin, epigenetic inheritance, histone ubiquitination, stress adaptation, fungal drug resistance