Clear Sky Science · en
Comparative analysis of deep mutational scanning datasets in enteroviruses A and B identifies functional divergence and therapeutic targets
Why tiny RNA viruses matter to us
Enteroviruses are a large group of tiny RNA viruses that quietly circulate around the globe, usually causing mild colds or stomach bugs but sometimes triggering paralysis, heart damage or other severe disease. We have few vaccines and even fewer drugs that work broadly against them, in part because these viruses evolve so quickly. This study asks a deceptively simple question with big practical stakes: when these viruses mutate, which parts of their machinery can change freely, and which parts are so vital that evolution keeps them almost frozen in place? The answers point toward new strategies for designing treatments that are hard for the virus to outsmart.
Reading the virus’s instruction book, one mutation at a time
The researchers focused on two human enteroviruses that cause very different illnesses: Enterovirus A71, linked to severe neurological disease in children, and coxsackievirus B3, associated with heart inflammation and even pancreatic cancer. Using a technique called deep mutational scanning, they created viral libraries in which nearly every position in each virus’s proteins was systematically changed. These mutant viruses were allowed to infect cells in culture, and high-throughput sequencing was used to measure how each change affected viral growth. By comparing each mutant to the original virus, the team built a detailed map of which sites in the viral proteome tolerate change and which are strongly constrained. 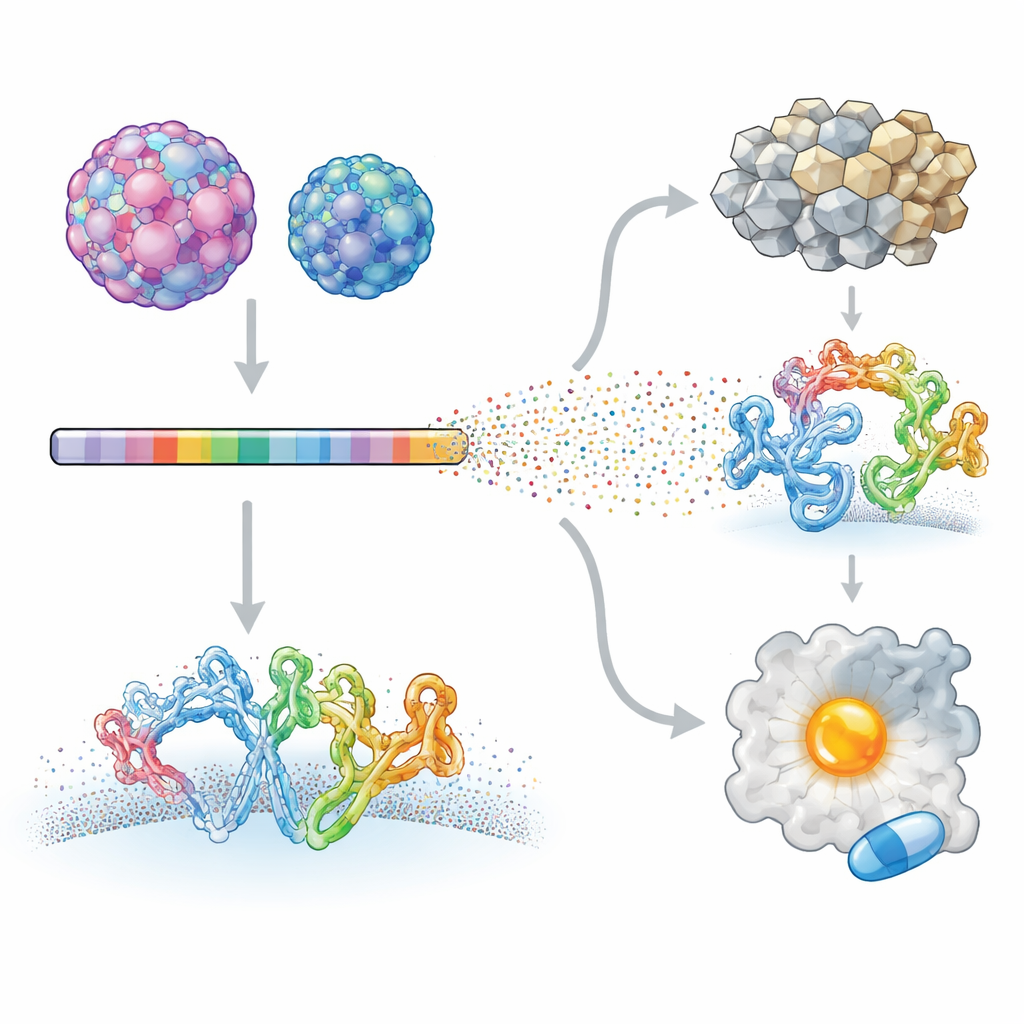
Shared hard limits and virus-specific flexibility
Despite sharing only about half of their amino-acid sequences, the two viruses showed strikingly similar overall patterns of constraint. The internal enzymatic “workhorses” that copy the genome, cut viral proteins and unwind RNA were highly sensitive to mutation in both viruses. Likewise, the hidden surfaces that hold together the protein shell, or capsid, proved difficult to alter without breaking the virus. In contrast, many outward-facing and host-interacting regions were more free to change and often differed sharply between the two species. Structural analysis showed that conserved, mutation-intolerant regions clustered around active sites and assembly interfaces, whereas regions that touch host receptors, membranes or immune molecules were hotspots of species-specific flexibility.
How viruses meet cells and dodge defenses
The team then zoomed in on the molecular handshake between virus and host. Enterovirus A71 and coxsackievirus B3 use different cellular receptors to gain entry, and the study found that the exact contact footprints on the capsid surface are among the most divergent regions in terms of mutation tolerance. Residues that grip the receptor for one virus are strongly constrained in that virus but relatively permissive in the other, reflecting how each has fine-tuned its docking platform. Similar divergence emerged in a small membrane-bound protein called 3A, which helps reshape cellular membranes and recruit host factors for genome replication. Modeling suggested that the two viruses use partly different contact patches on 3A to engage the same host factor, GBF1, and may also insert this protein into membranes at different depths. These differences help explain why closely related viruses can infect different tissues and provoke distinct disease patterns.
What lab evolution misses—and what it gets right
To place their cell-culture experiments in real-world context, the authors compared their mutation maps with thousands of natural virus sequences from patient samples. Overall, sites that were flexible in the lab tended also to be variable in nature, especially across broader species-level comparisons. However, when they examined which specific amino acids were preferred at each site, natural evolution and lab selection agreed most closely within a single virus type. Notably, the greatest mismatches clustered on the outer capsid surface and on host-interacting regions of non-structural proteins 2A and 3A—precisely where immune responses and complex host environments are expected to matter most. This suggests that deep mutational scanning captures the basic mechanical constraints of viral proteins, while real infections overlay additional pressures from immunity and tissue tropism that are harder to reproduce in vitro.
Finding a shared weak spot for future drugs
Finally, the researchers asked whether these maps could highlight a common Achilles’ heel for drug development. Using computational tools, they searched both viruses for pockets on protein surfaces that could, in principle, bind small-molecule drugs. They then overlaid the mutational data to see which pockets were made of residues that the virus cannot easily change without harming itself. One pocket, located on the 2C helicase—a ring-shaped motor that helps unwind the viral RNA—stood out. Its shape and constituent residues are highly conserved across four major human enterovirus species, it sits away from already-known active sites, and changes there are both rare in nature and strongly detrimental in lab tests. 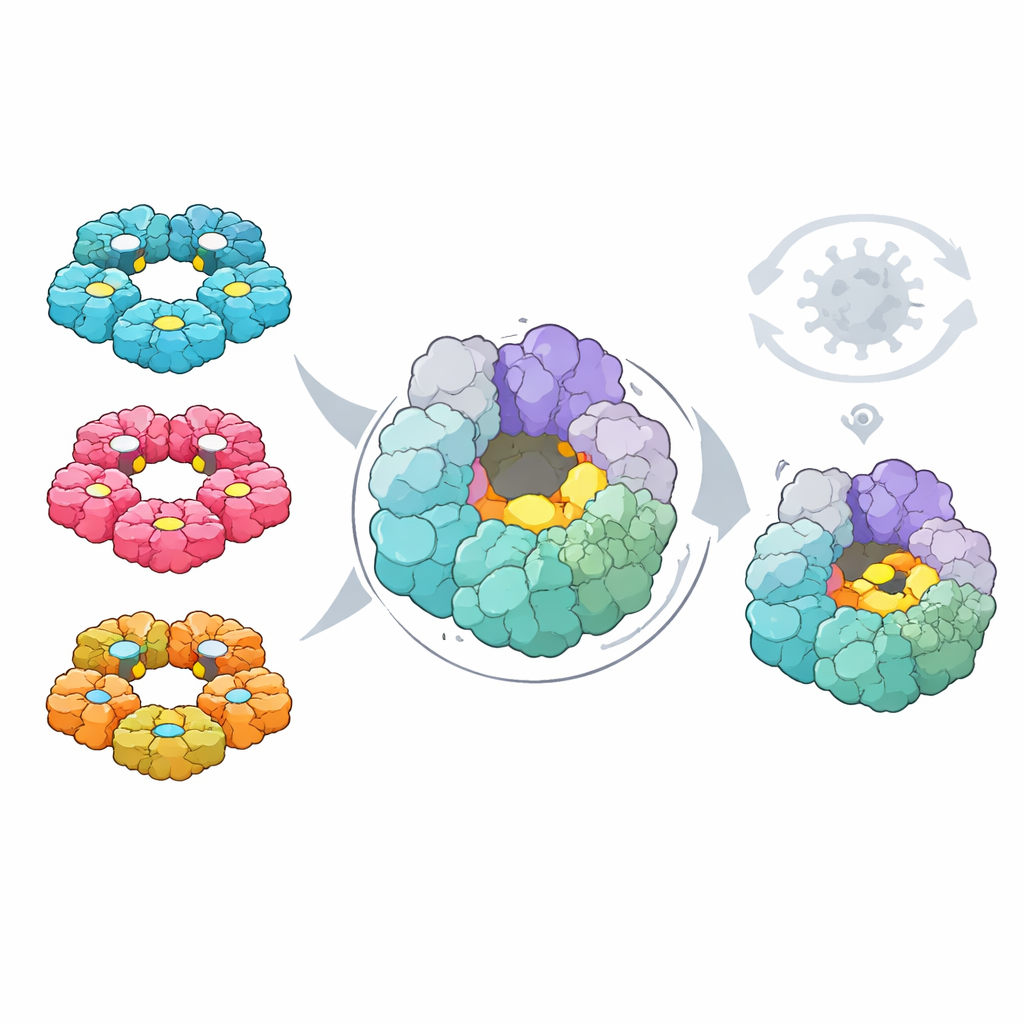
What this means for future treatments
By systematically probing how every possible single-letter change affects two important enteroviruses, this study reveals a clear division between a shared, rigid core of viral machinery and more flexible, virus-specific interfaces with the host. The conserved core includes a newly highlighted pocket on the 2C helicase that appears difficult for the virus to mutate without sacrificing fitness, making it an attractive target for broad-spectrum antivirals with a high barrier to resistance. At the same time, the more adaptable outer surfaces and host-contact sites explain why related viruses behave so differently in the body and point to where vaccines and immune-based therapies must contend with rapid evolution. Together, these findings provide a roadmap for designing treatments that exploit the virus’s own evolutionary limits.
Citation: Álvarez-Rodríguez, B., Bakhache, W., McCormick, L. et al. Comparative analysis of deep mutational scanning datasets in enteroviruses A and B identifies functional divergence and therapeutic targets. Nat Ecol Evol 10, 467–480 (2026). https://doi.org/10.1038/s41559-026-02993-8
Keywords: enteroviruses, viral evolution, deep mutational scanning, antiviral targets, 2C helicase