Clear Sky Science · en
Dynamical network analysis reveals long-range residue couplings at the pMHC interface underlying enhanced immunogenicity
How Tiny Viral Fragments Steer Our Immune Defenses
Our killer T cells patrol the body looking for signs of infection or cancer. They do this by scanning tiny protein fragments, called peptides, displayed on cell surfaces by molecules known as MHC class I. This study asks a subtle but important question: how can a single small change in one of these peptides make T cells respond much more strongly—or not at all? The answer turns out to involve not just static structure, but how the whole molecular assembly moves and flexes over time.
The Lock, the Key, and the Moving Parts
To understand the work, it helps to picture the peptide–MHC (pMHC) complex as a lock and the T cell receptor (TCR) as a key. The peptide sits in a groove on the MHC molecule, and together they form the surface that the TCR probes. Previous research has shown that both the exact peptide sequence and the particular MHC variant strongly influence whether a T cell responds. Scientists have also engineered “altered peptide ligands” that carry small changes to tune immune responses, including in cancer immunotherapy. But while we know a lot about the static shapes of these complexes, we know far less about how motions at one point in the peptide can affect distant parts of the interface where the TCR actually binds.
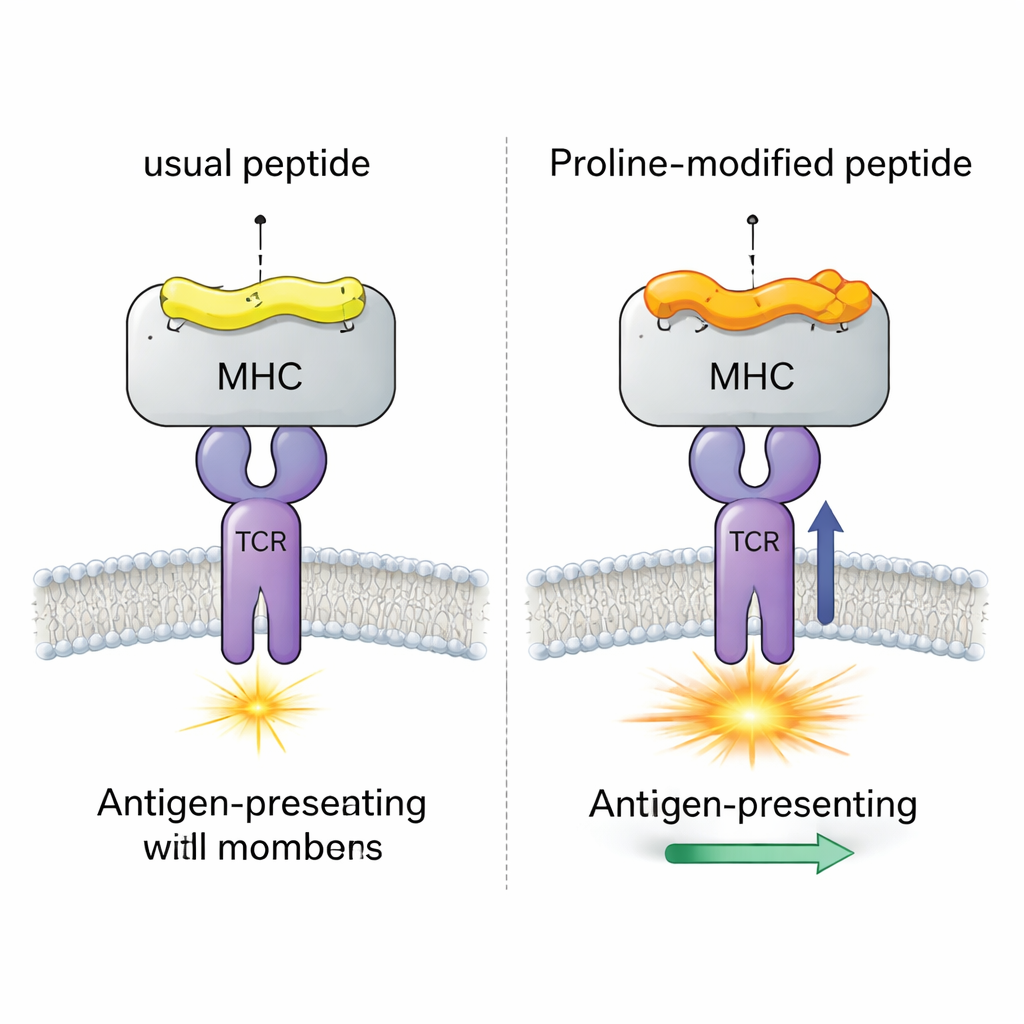
A Viral Test Case with Four Nearly Identical Peptides
The team focused on a well-studied mouse virus (LCMV) system involving the peptide gp33, which normally triggers strong CD8+ T cell responses. They compared four closely related versions of this peptide, all bound to the same MHC molecule (H-2Db). One version is the original viral peptide; one carries an immune-escape mutation that T cells barely recognize; and two are “proline-altered” vaccine candidates where a single amino acid near the start of the peptide is changed to proline. Earlier experiments showed that this proline swap increases how tightly the peptide–MHC complex holds together and how strongly a model TCR (called P14) responds, but the detailed mechanism remained unclear.
Watching Molecules Jiggle: Simulations Meet Crystallography
To uncover what is happening, the authors combined high-resolution crystal structures with long, atom-by-atom computer simulations of each pMHC complex in motion. They examined how much each amino acid residue fluctuates over time and how these fluctuations change when the peptide’s third position is converted to proline. By correlating motion patterns across many paired simulations, they built a “dynamic map” of which residues move together, even when they are far apart in space. They then turned this map into a network, where each residue is a node and edges represent statistically linked motions, and analyzed this network using graph-theory tools similar to those used in social network analysis.
Long-Distance Communication Inside the Immune Lock
The central finding is that altering the third peptide residue to proline does more than simply stiffen that local spot. It changes how motion is transmitted along one of the MHC helices that borders the peptide-binding groove. This in turn affects the behavior of another peptide residue, position six, which sits right under the TCR’s footprint and is critical for recognition. In the “good” proline-modified versions, this residue samples a wider range of conformations, including those that are optimal for TCR binding. In the immune-escape variant without proline, that residue is more locked in place and rarely adopts the TCR-friendly orientation. Network analysis reveals that this influence travels via specific amino acids in the MHC groove, forming a chain of dynamically coupled residues that link the site of the proline change to the TCR-contact region.
Why This Matters for Vaccines and Immunotherapy
These results show that immunogenicity—how strongly a peptide triggers T cells—is not just about whether the shapes fit at a single moment, but also about how the complex breathes and flexes over time. A subtle change in one position can ripple through the molecular network, making key contact residues more likely to present themselves in TCR-compatible poses. The authors’ computational workflow offers a way to detect such long-range couplings systematically, which could help guide the design of altered peptides for vaccines and cancer therapies. In simple terms, they demonstrate that by carefully choosing where to tweak a peptide, we can nudge the whole lock into a more “ready-to-be-opened” dynamic state for the immune system’s key.
Citation: Resink, T., Sala, B.M., Sun, R. et al. Dynamical network analysis reveals long-range residue couplings at the pMHC interface underlying enhanced immunogenicity. npj Syst Biol Appl 12, 15 (2026). https://doi.org/10.1038/s41540-026-00653-y
Keywords: T cell recognition, peptide MHC, protein dynamics, altered peptide ligands, immunogenicity