Clear Sky Science · en
The critical role of intrinsic defects and many-body interactions on the stability of MnBi2Te4
Why tiny flaws in crystals matter for future tech
Many of tomorrow’s quantum technologies—such as ultra-efficient electronics and new kinds of computers—rely on exotic materials whose surfaces conduct electricity while their interiors remain insulating. One of the most promising of these is MnBi2Te4, a “topological magnet” that could host resistance-free edge currents useful for low-power devices and quantum computing. But in real crystals, atoms often sit in the wrong places, and these tiny flaws can quietly destroy the very effects engineers want to use. This study asks a basic but crucial question: are those flaws a manufacturing accident, or are they actually favored by nature at the temperatures where the material is made?
A promising material with a stubborn problem
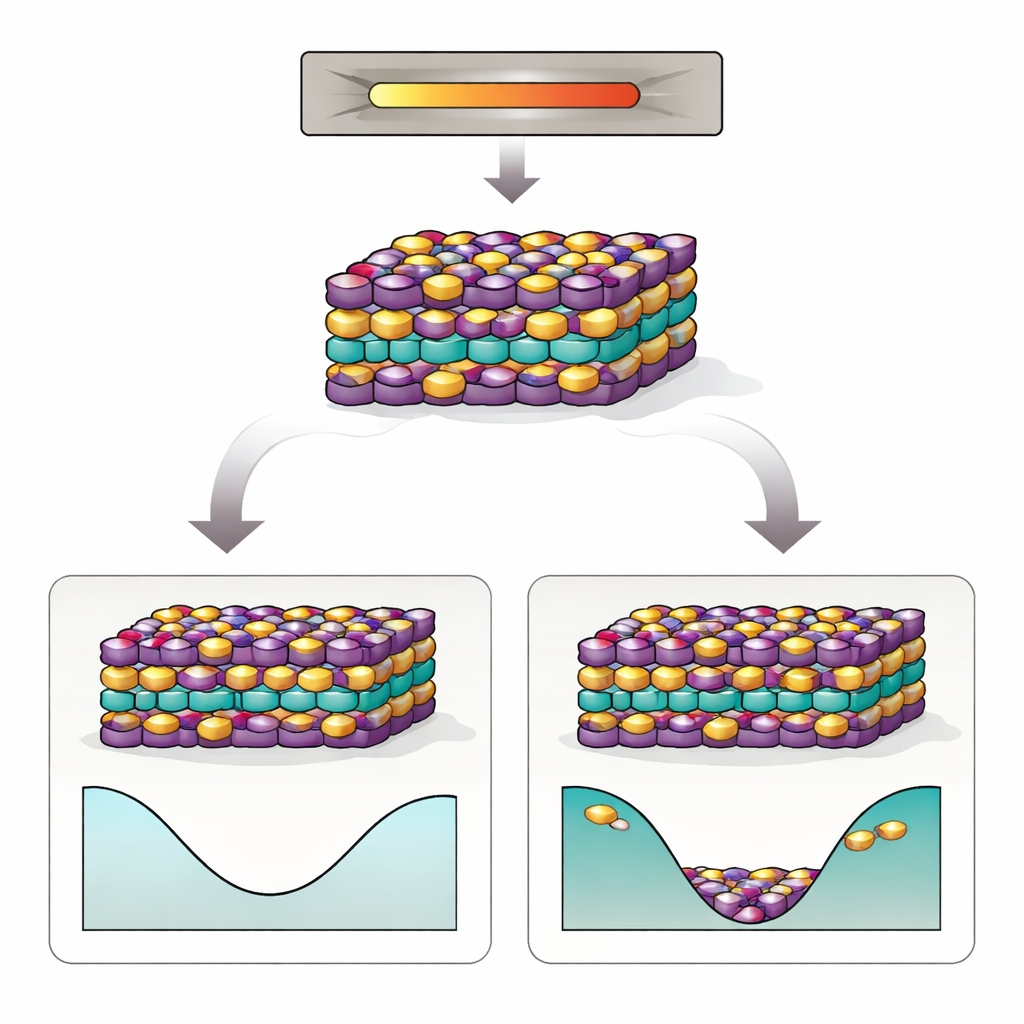
MnBi2Te4 is built from stacked atomic sheets, like a carefully ordered club sandwich. Its special electronic behavior depends on two things: a precise arrangement of manganese (Mn), bismuth (Bi), and tellurium (Te) atoms, and a delicate pattern of magnetic alignment between the layers. Experiments, however, repeatedly find that many Mn and Bi atoms swap places—so‑called antisite defects. These swaps scramble the magnetic pattern, push the material away from its ideal insulating state, and make it harder to observe the sought‑after quantum phenomena. Worse, even when crystals are grown and annealed with great care, the antisite defects stubbornly remain, suggesting that something deeper than imperfect processing is at work.
Why earlier calculations disagreed with experiments
Standard computer simulations had painted a puzzling picture. At absolute zero, common quantum‑mechanical methods predicted that creating a Mn–Bi swap costs energy and should therefore be rare. That clashes with experiments showing high defect levels in real samples produced around 850 kelvin (over 500 °C). The authors argue that two key pieces were missing from earlier theory. First, defects were usually treated one at a time, ignoring how they interact and cluster. Second, calculations were typically done at zero temperature, neglecting the way heat and disorder change which atomic arrangements are favored. In a material that is only marginally stable to begin with, even small contributions from the “many‑body” behavior of electrons and from the sheer number of possible arrangements can tip the balance.
Following every swap in a virtual crystal
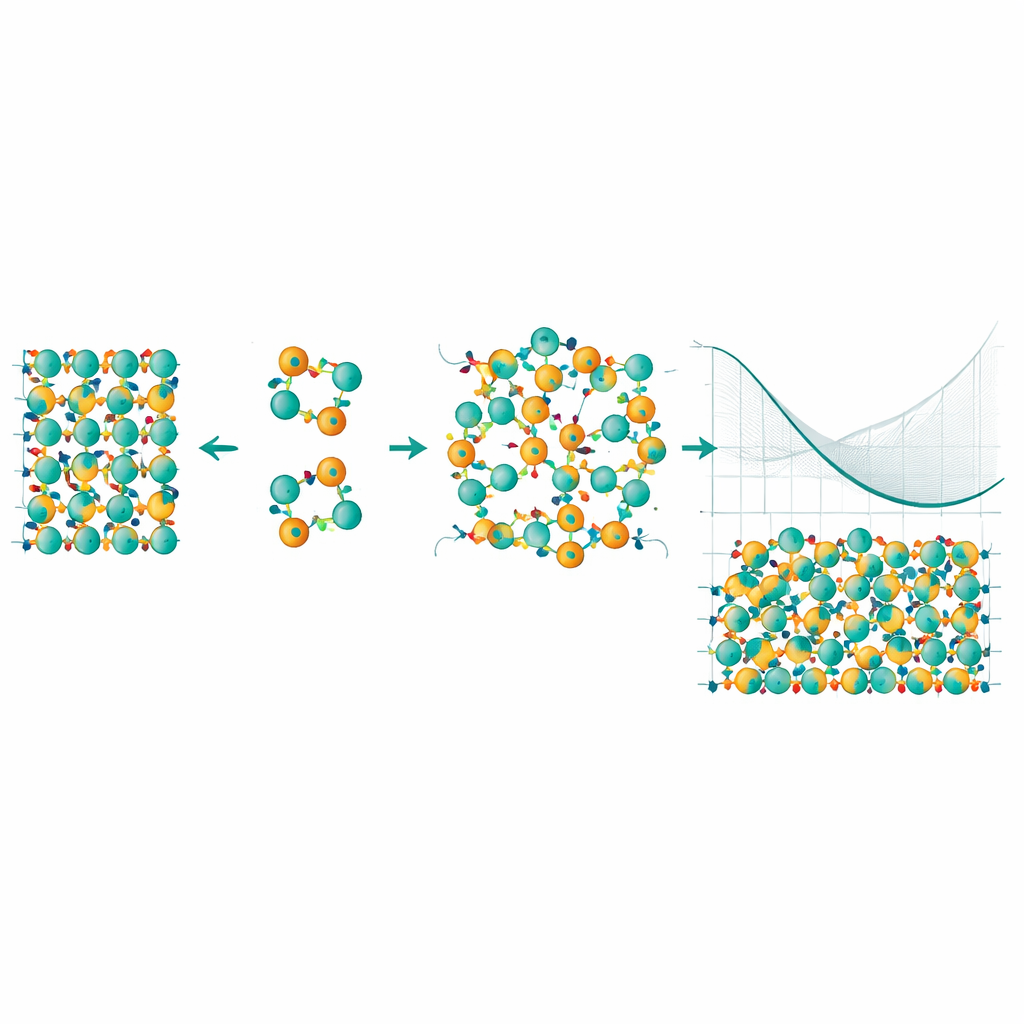
To tackle this, the researchers built a statistical model that can explore millions of different ways Mn and Bi atoms might rearrange. They used a technique called cluster expansion, which breaks the crystal’s energy into contributions from single atoms, pairs, and small groups, and then combined it with Monte Carlo sampling to see which patterns appear at different temperatures. Crucially, they corrected the underlying energies using an especially accurate method known as quantum Monte Carlo, which better captures subtle electron–electron interactions. This hybrid approach let them calculate not just the energy cost of a single swap, but how that cost changes as more defects appear and begin to influence one another.
When disorder becomes the cheaper option
The simulations reveal that interactions between multiple antisite defects and the “configurational entropy” of disorder—essentially the huge number of ways to arrange the swapped atoms—dramatically reshape the material’s behavior at growth temperatures. Although a lone Mn–Bi swap is costly at zero temperature, at higher temperatures the gain in entropy outweighs this energy cost. The authors find an order–disorder transition near the synthesis temperature: above this point, swapped Mn and Bi atoms become thermodynamically favored, and the free energy of a defective crystal actually drops below that of a perfectly ordered one. In other words, nature prefers a crystal with a substantial fraction of antisite defects, and these defects tend to form in correlated clusters rather than appearing randomly.
What this means for making better quantum materials
For non‑experts, the main takeaway is that the troublesome defects in MnBi2Te4 are not simply a manufacturing flaw; they are a natural consequence of the material’s thermodynamics at the temperatures where it is grown. The study shows that once many‑body interactions and the statistics of disorder are properly included, theory and experiment finally agree: antisite defects form spontaneously and in large numbers. This insight explains why producing truly defect‑free crystals has been so difficult, and it offers a roadmap for improving other delicate quantum materials. Any effort to engineer better samples—by changing growth conditions, compositions, or processing routes—must contend with the fact that, at high temperature, disorder is not an accident but the lowest‑energy choice for the crystal.
Citation: Ghaffar, A., Saritas, K. & Reboredo, F.A. The critical role of intrinsic defects and many-body interactions on the stability of MnBi2Te4. npj Comput Mater 12, 119 (2026). https://doi.org/10.1038/s41524-026-02019-8
Keywords: topological insulators, magnetic materials, crystal defects, quantum Monte Carlo, materials thermodynamics