Clear Sky Science · en
Active learning potentials for first-principles phase diagrams using replica-exchange nested sampling
Why this matters for future materials
From faster computer chips to tougher airplane parts, many modern technologies depend on knowing how a material changes when it is heated up or squeezed under pressure. These changes, called phase transitions, are summarized in phase diagrams—the maps that tell scientists which form of a material is stable under which conditions. This study introduces a new way to automatically draw such maps directly from quantum‑mechanical calculations, using artificial intelligence to dramatically cut the cost while keeping high accuracy.
Mapping materials without guesswork
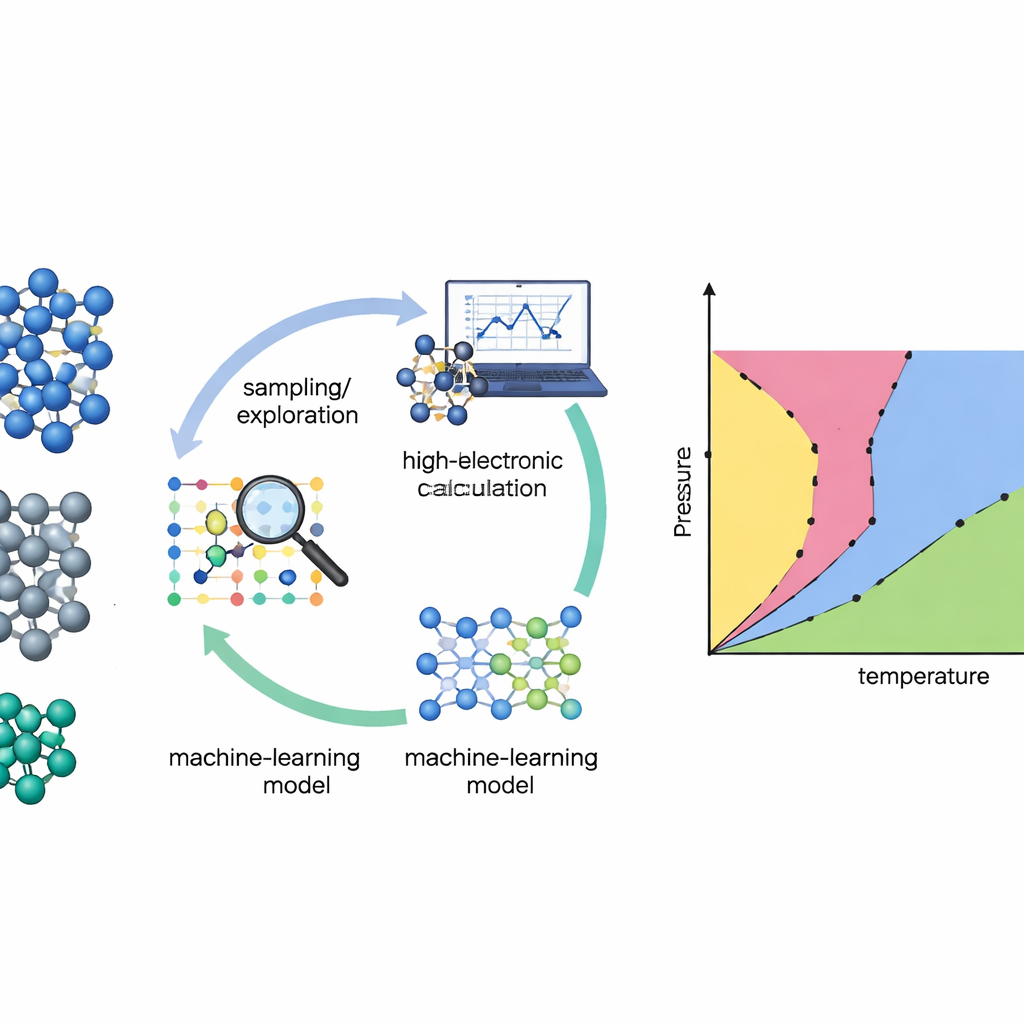
Traditionally, building a phase diagram from first principles is like hiking through a rugged landscape in the dark: you must already suspect where the important valleys and mountain passes are. Many standard methods work only if researchers feed in strong prior knowledge about which crystal structures or “paths” to explore. The authors instead rely on a technique called nested sampling, which systematically combs through the full energy landscape of a material without assuming which phases will appear. By tracking how accessible different regions of that landscape are, nested sampling can recover thermodynamic properties and phase changes over a wide range of temperatures in a single sweep.
Letting the model choose what it needs to learn
Even the smartest search method needs a good description of how atoms interact. Direct quantum‑mechanical calculations (density functional theory) are accurate but too expensive to evaluate millions or billions of times. The team tackles this by training machine‑learning interatomic potentials—fast models that mimic the quantum forces between atoms. The catch is that such models are only trustworthy where they have seen enough examples. To solve this, the authors build an active‑learning loop: the machine‑learning model runs the nested sampling simulation, flags configurations where it is uncertain, and then asks for high‑level quantum calculations only on this carefully chosen subset. The new data are fed back into the model, which becomes more reliable in the regions that matter most for the phase diagram.
A new engine for exploring silicon, germanium, and titanium
The researchers tested their approach on three important elements: silicon and germanium, well‑known semiconductors, and titanium, a widely used structural metal. They began from modest initial databases built from known crystal structures and simple distortions, deliberately omitting liquids and many high‑energy arrangements. Replica‑exchange nested sampling—many nested sampling runs at different pressures that can swap configurations—then explored the materials’ energy landscapes. After each exploration round, the algorithm automatically selected hundreds of representative atomic configurations, weighted toward those where its force predictions disagreed most across a committee of neural‑network models. These were recomputed with a high‑accuracy quantum method (r2SCAN) and used to retrain the potentials before launching the next round.
From noisy beginnings to reliable phase maps
Over about ten to fifteen learning cycles, the models’ uncertainty steadily shrank, especially in the forces that govern atomic motion. At the same time, the nested sampling trajectories began to reveal the familiar outlines of the phase diagrams. For silicon, the method reproduced the known low‑pressure diamond structure, its high‑pressure hexagonal phase, and the characteristic melting behavior with temperature and pressure, all in good agreement with experiments and earlier simulations. Germanium showed a similar pattern, with a low‑pressure diamond‑like phase giving way to a high‑pressure metallic phase, though the exact transition pressure shifted somewhat because of the chosen quantum‑mechanical approximation. Titanium provided a tougher test: its phases are metallic, structurally similar, and separated by small energy differences. Even there, the active‑learning strategy captured the sequence of solid phases and the melting line, and additional checks using radial distribution functions confirmed the identities of the predicted structures.
What this means for designing new materials
In plain terms, the study shows that a computer can now teach itself how a material behaves across a broad range of temperatures and pressures, asking a quantum‑mechanical “oracle” only when necessary. The replica‑exchange nested sampling engine guarantees broad and unbiased exploration, while the active‑learning loop ensures that the machine‑learning potentials are accurate where it counts thermodynamically. Although the current work focuses on three elements and one particular quantum method, the framework is general: it can be paired with more advanced electronic theories or powerful neural networks, and extended to complex alloys or compounds. As computing power and algorithms improve, this kind of autonomous workflow could become a standard tool for predicting phase diagrams and guiding the discovery of new materials with tailored properties.
Citation: Unglert, N., Ketter, M. & Madsen, G.K.H. Active learning potentials for first-principles phase diagrams using replica-exchange nested sampling. npj Comput Mater 12, 107 (2026). https://doi.org/10.1038/s41524-026-01989-z
Keywords: materials phase diagrams, active learning, machine learning potentials, nested sampling, silicon germanium titanium