Clear Sky Science · en
Optical properties of a diamond NV color center from capped embedded multiconfigurational correlated wavefunction theory
Diamonds as Tiny Quantum Light Switches
Most people know diamond for its sparkle, but inside its crystal lattice, tiny imperfections can act as powerful building blocks for future quantum computers and sensors. One such imperfection, called a nitrogen-vacancy (NV) center, can store and process quantum information using the spin of just a few electrons. This paper shows how a new form of advanced computer simulation can predict the light‑absorption and emission behavior of this diamond defect with remarkable accuracy, helping scientists design better quantum devices from the atom up.
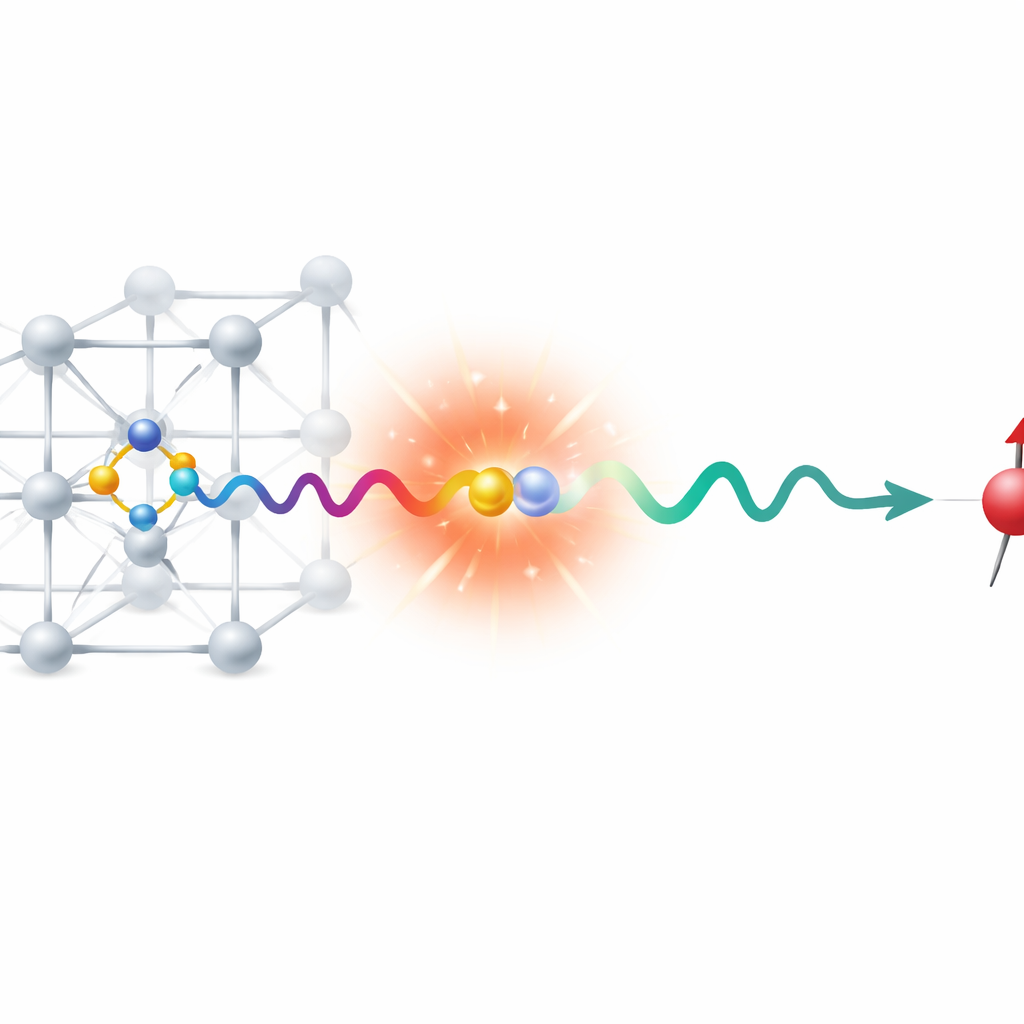
A Special Flaw in an Otherwise Perfect Gem
In a perfect diamond, each carbon atom bonds neatly to four neighbors in a rigid three‑dimensional network. An NV center appears when one carbon is replaced by a nitrogen atom and a neighboring site is left empty—a vacancy. This rearrangement leaves three nearby carbon atoms with “dangling” bonds that each carry an unpaired electron. When the defect gains an extra electron overall, two of these electrons remain unpaired, giving the center a spin triplet ground state. Light can promote one of these electrons to a higher‑energy orbital, and when it falls back, the defect glows. Because the energies of these jumps lie in the visible and infrared, well below diamond’s own deep‑ultraviolet band gap, the NV center behaves as a bright color center embedded in an otherwise transparent host.
From Light and Magnetism to Quantum Bits
The NV center’s usefulness stems from the fact that its electron spins can be treated as quantum bits, or qubits. Different spin orientations act like the logical “0” and “1,” but—unlike ordinary bits—they can exist in combinations of both at once. In a magnetic field, the three spin levels of the triplet state split in energy, and microwave radiation can drive transitions between them. At the same time, visible light excites the defect, and the brightness of its fluorescence depends on which spin state it occupies. This spin‑dependent glow lets researchers read out the qubit optically. However, unwanted pathways exist: the excited triplet can relax into spin‑singlet states through intersystem crossing, temporarily shelving the defect in a non‑magnetic state and altering its brightness. Predicting the exact energies of all these triplet and singlet levels, and the gaps between them, is crucial for controlling NV‑based devices.
Why Ordinary Calculations Fall Short
Most large‑scale computer studies of solids use density functional theory (DFT), which represents electrons in terms of an effective average field. While efficient, standard DFT struggles with situations where several electronic configurations contribute strongly at once—exactly the case for the NV center’s singlet states. It also tends to misplace the energies of defect levels relative to the host crystal’s bands. More rigorous “multireference” wavefunction methods can treat these subtleties, but they are far too expensive to apply directly to a realistic chunk of diamond containing many atoms. Previous high‑accuracy approaches have therefore relied on huge periodic supercells or elaborate embedding schemes, often at great computational cost and with mixed success in reproducing experimental excitation energies.
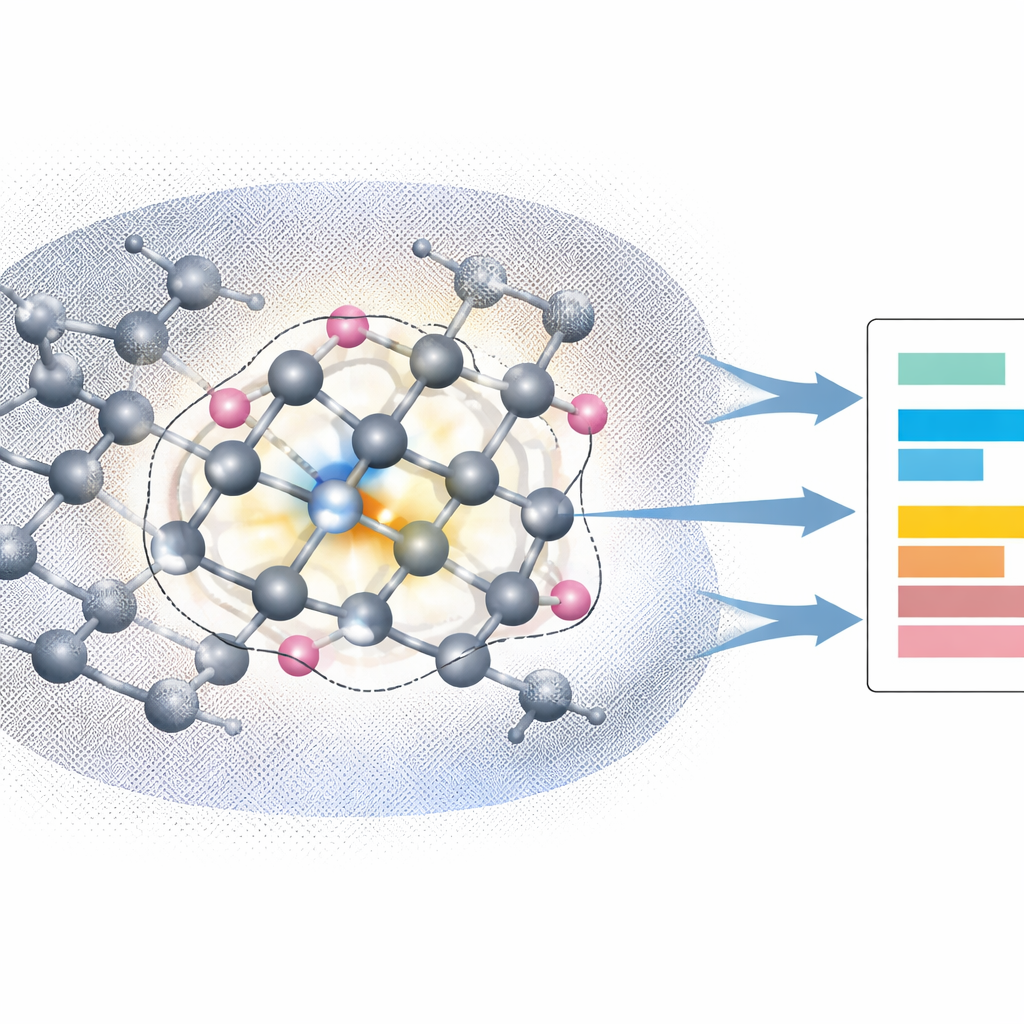
Zooming In on the Defect with Capped Embedding
The author tackles this challenge with a technique called capped density functional embedding theory (capped‑DFET). The idea is to carve out a small cluster of atoms around the NV center—just the nitrogen, the three adjacent carbons, and their nearest neighbors—and surround the cut bonds with carefully chosen “capping” atoms that mimic the missing parts of the crystal. The rest of the diamond is treated at the DFT level and folded into an effective local potential that acts on the cluster. This potential is adjusted so that, taken together, the cluster plus environment reproduce the electron density of the full solid. Within this embedded cluster, the study then applies a high‑end multiconfigurational method (CASSCF with NEVPT2 corrections) that explicitly accounts for all important electron rearrangements in both triplet and singlet states.
Reaching Experimental Accuracy with a Tiny Model
Using this embedded cluster, the calculations reproduce the measured vertical excitation energies of the NV center’s key optical transitions to within about 0.1 electron‑volt, for both the bright triplet transition and the infrared singlet transition. They also match the inferred energy gap that controls intersystem crossing between an excited triplet and an excited singlet. Notably, the predicted excitation energies hardly change when the surrounding periodic diamond cell is enlarged, and they depend only weakly on how large the embedded cluster is, as long as it includes the defect and its closest neighbors. This shows that the capped‑DFET approach captures the local physics of the NV center while avoiding spurious long‑range interactions between periodically repeated charged defects.
What This Means for Future Quantum Materials
In plain terms, this work demonstrates that a relatively small, carefully embedded fragment of diamond can stand in for a much larger crystal when simulating an NV center’s optical and magnetic behavior. The method delivers near‑experimental precision for the energies that govern how the defect absorbs and emits light and how its spin states interconvert—properties that directly affect how well it can serve as a qubit or nanoscale sensor. Because the approach is both accurate and computationally efficient, it can now be applied to explore new defects and host materials, guiding the search for the next generation of solid‑state quantum technologies.
Citation: Martirez, J.M.P. Optical properties of a diamond NV color center from capped embedded multiconfigurational correlated wavefunction theory. npj Comput Mater 12, 113 (2026). https://doi.org/10.1038/s41524-026-01987-1
Keywords: nitrogen vacancy center, diamond qubits, quantum defects, electronic structure theory, computational materials