Clear Sky Science · en
Efficient and accurate spatial mixing of machine learned interatomic potentials for materials science
Why faster atomic simulations matter
Designing better materials for technologies like nuclear fusion, microelectronics, and structural alloys increasingly relies on computer simulations that track how atoms move and interact. The most accurate methods borrow ideas from quantum physics, but they are so computationally demanding that only modest system sizes and timescales are practical. This article introduces ML‑MIX, a technique and software package that lets researchers keep near–quantum accuracy exactly where it is needed, while using simpler, cheaper models everywhere else. The result is a substantial speed boost—often a factor of 4 to 10—without losing reliability in the key physical predictions.
Blending detailed and simple views of atoms
At the heart of the work is a simple idea: not every atom in a simulation needs the same level of attention. Regions where bonds are stretching, breaking, or rearranging—such as defects, surfaces, or implanted particles—benefit from modern machine‑learning interatomic potentials, which mimic quantum‑mechanical accuracy. But atoms far away from these “hot spots” mostly vibrate around regular positions and can be handled by much simpler models. ML‑MIX provides a way to combine an accurate but expensive model with a leaner “cheap” one inside the same simulation box. It does this by defining a core zone that uses the expensive model, a surrounding buffer where forces are carefully blended between models, and an outer bulk zone that uses only the cheap description. 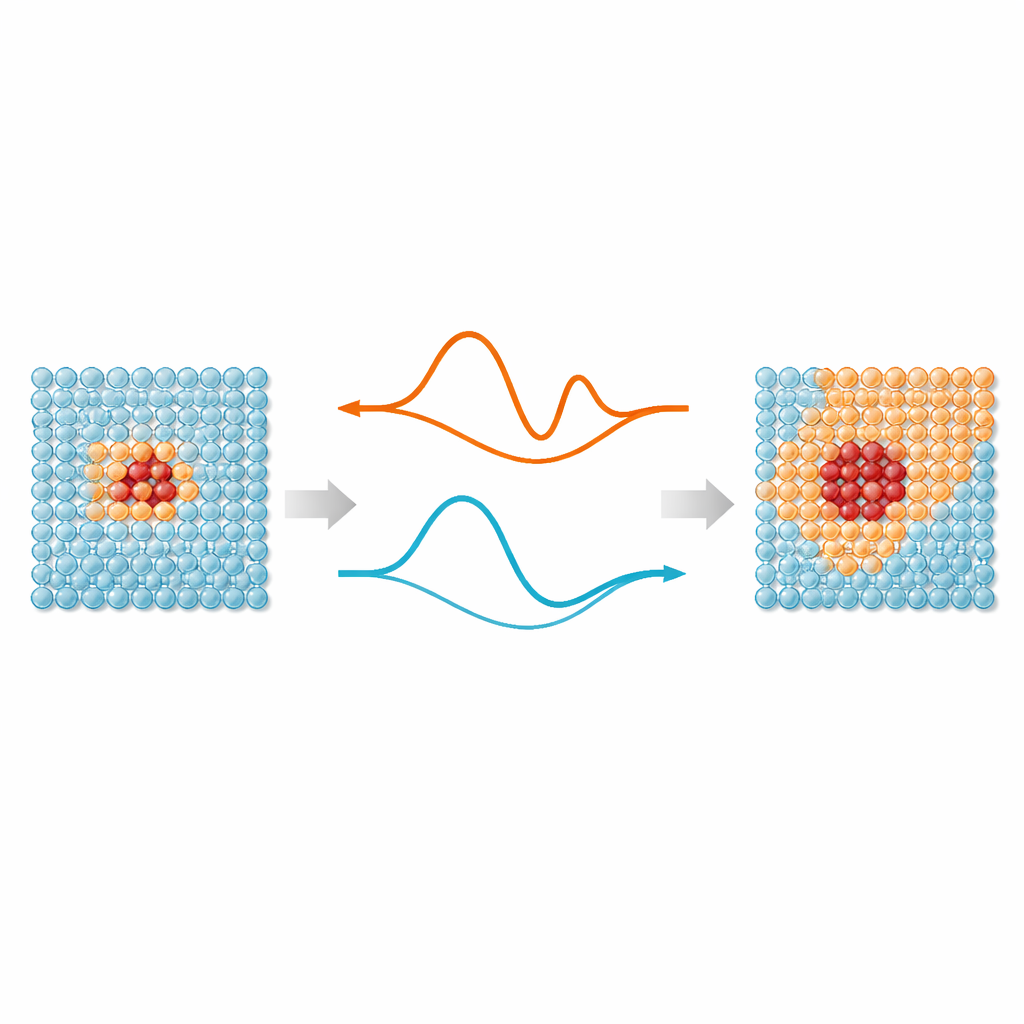
Teaching a cheap model to mimic an accurate one
A key challenge is making sure the cheap model behaves like the accurate one wherever they meet. Rather than fitting the cheap model directly to a vast and varied quantum‑mechanical dataset, the authors generate focused “synthetic” data by running the accurate model in the specific conditions relevant to the bulk region: high‑temperature vibrations and gently strained crystals. They then fit the cheap model so that it matches this data, while imposing strict constraints on basic material properties such as the elastic constants and lattice spacing. This constrained fitting ensures that long‑range stresses and strains match smoothly across the boundary between the two models, avoiding artificial forces that could corrupt the dynamics near the interface.
Putting the method to the test
To check that ML‑MIX really works, the authors run a suite of tests on silicon, iron, and tungsten systems. For a simple example, they compute the energy barrier for a vacancy—an empty lattice site—in silicon to move from one position to another. The mixed simulation reproduces the result of an all‑expensive calculation to within a thousandth of an electron‑volt, while running about five times faster. In a more dynamic setting, they stretch a single silicon bond in a hot crystal and measure the average force on it. A simulation that uses only the cheap model already comes surprisingly close, but once a small expensive core is added around the stretched bond, the agreement becomes statistically indistinguishable from the fully accurate reference, with speedups of up to a factor of about 13 in serial runs.
Following defects and particles in motion
More realistic tests examine how defects move through metals. The team simulates the diffusion of a self‑interstitial defect in iron and of helium atoms inside tungsten. In each case, the expensive model is confined to a small moving region around the defect, while the rest of the crystal is handled by the cheap potential. The resulting diffusion coefficients match those from fully accurate simulations within statistical error, even when a cheap‑only simulation would fail. The authors then push the method to larger, scientifically important problems in tungsten, a leading candidate material for fusion reactors. They model the motion of screw dislocations—line‑like defects that control plastic deformation—and the implantation of helium atoms into a hot tungsten surface. In both cases, ML‑MIX reproduces the expensive‑only results while cutting the computational cost by factors of about four to eleven. 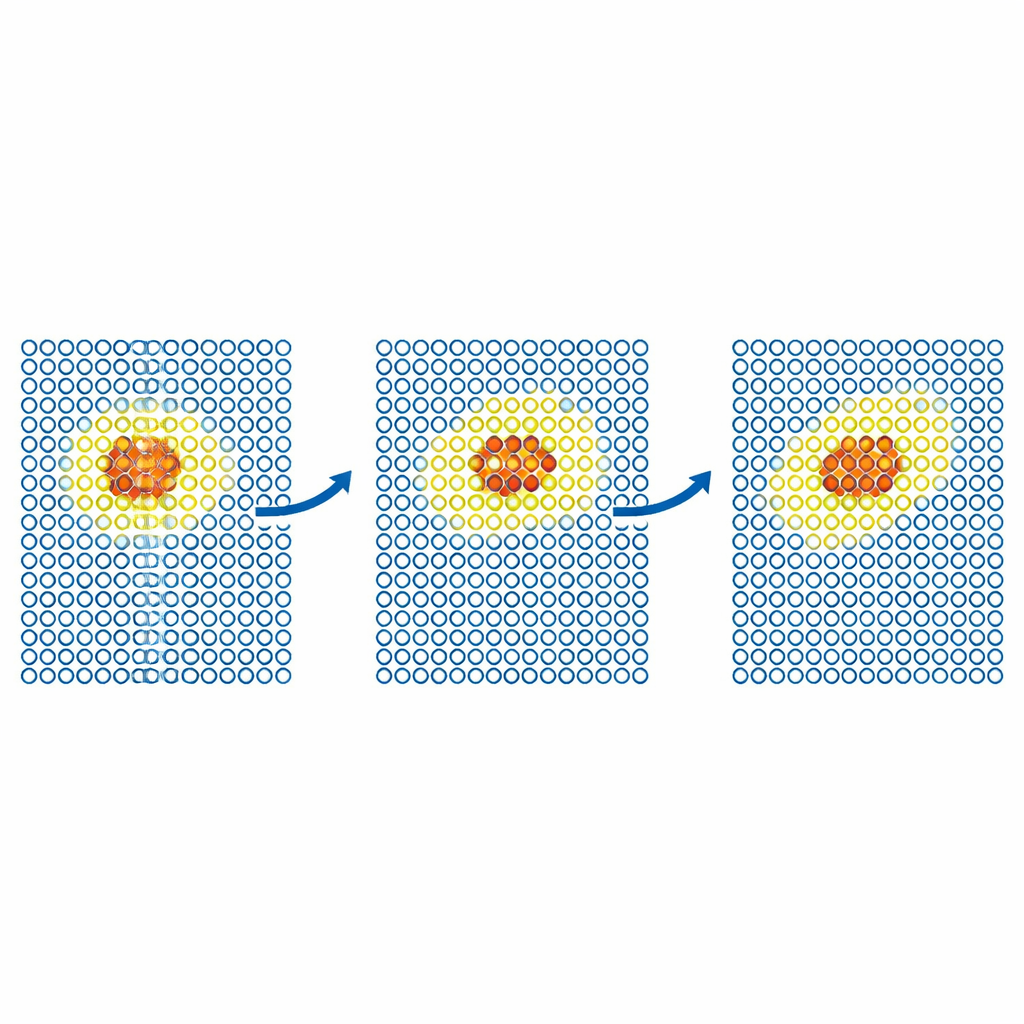
Matching experiments and looking ahead
The helium implantation study shows the power of this approach most clearly. By mixing a cutting‑edge machine‑learning model for helium–tungsten interactions with a faster potential for pure tungsten, the authors simulate many more impact events and larger samples than would otherwise be feasible, all on graphics processors. The predicted fraction of helium atoms that bounce off the surface versus implanting inside the metal agrees with experimental measurements up to incident energies of about 80 electron‑volts, something earlier simulations struggled to achieve. Although the mixing scheme does not strictly conserve energy and requires gentle thermostats, the resulting drift is small and manageable. Overall, ML‑MIX demonstrates that carefully combining detailed and simplified atomic models can break long‑standing barriers between accuracy and scale, opening the door to routine, high‑fidelity simulations of complex materials in realistic environments.
Citation: Birks, F., Nutter, M., Swinburne, T.D. et al. Efficient and accurate spatial mixing of machine learned interatomic potentials for materials science. npj Comput Mater 12, 110 (2026). https://doi.org/10.1038/s41524-026-01982-6
Keywords: machine learned interatomic potentials, multiscale materials simulation, tungsten helium implantation, defects and dislocations, molecular dynamics acceleration