Clear Sky Science · en
Graph atomic cluster expansion for foundational machine learning interatomic potentials
Teaching Computers to Feel the Atoms
Designing new materials for batteries, airplanes, or fusion reactors often comes down to a simple question: how do atoms push and pull on each other? Computing these forces exactly is so expensive that it can take days on a supercomputer for a single material. This paper introduces a new family of machine-learning models, called GRACE, that act like a universal “calculator” for atomic forces across most of the periodic table. They aim to make accurate simulations of complex materials routine rather than heroic.
A Single Model for Many Materials
Most existing machine-learning force fields are specialist tools: they work very well for a few elements or compounds, but must be rebuilt from scratch when new elements are added. GRACE takes a different route. It is designed from the start as a foundational model that can handle 89 chemical elements and an enormous variety of atomic arrangements with one shared set of rules. To do this, the authors build on a mathematical framework called the atomic cluster expansion and extend it to graph-like structures, allowing the model to describe both local neighborhoods of atoms and more extended patterns in a unified way. Instead of hard-coding every possible interaction, GRACE learns compact “embeddings” that capture similarities among elements, so that knowledge about one material can help describe another.
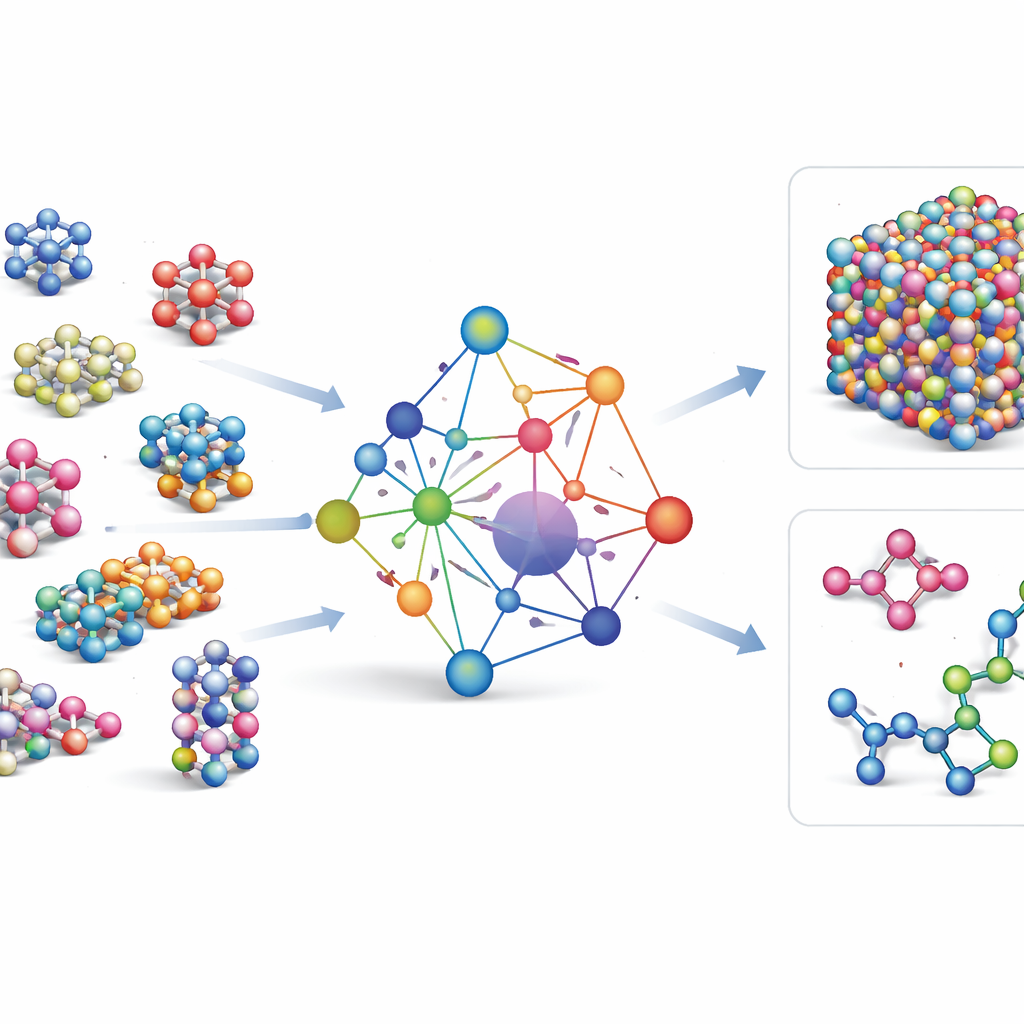
Training on a Sea of Atomic Data
To teach GRACE how atoms behave, the authors assembled some of the largest public databases of quantum-mechanical calculations. The core is the OMat24 collection, which contains around 110 million simulations of inorganic materials, supplemented by two others that track how structures relax and evolve. Together, these datasets cover near-equilibrium crystals, strained and distorted structures, high-temperature liquids, and more, across the same broad set of elements. GRACE models come in several sizes, from simpler one-layer versions that look only at local atomic environments to deeper two-layer versions that effectively pass “messages” between neighboring regions. Initial training aims for a good balance of energies, forces, and internal stresses, and further fine-tuning adjusts the models to be compatible with widely used reference databases in materials science.
Putting the Model to the Test
A universal model is only useful if it performs reliably across many tasks. The authors therefore put GRACE through a demanding test suite that mirrors how scientists actually use atomistic simulations. On a community benchmark for discovering stable crystal structures, GRACE consistently sits on the “Pareto front”: for a given accuracy, it is faster than competing models, and for a given speed, it is more accurate. Similar advantages appear when predicting thermal conductivity, a property that depends sensitively on tiny changes in atomic motion. GRACE also does well on elastic properties, surface energies, grain boundary energies, and point defect formation energies in many pure metals, all of which probe how materials respond to being stretched, cut, or locally damaged. A long molecular-dynamics run of a hot molten salt shows that the model remains numerically stable for nanoseconds while reproducing detailed structural patterns and atomic diffusion rates.
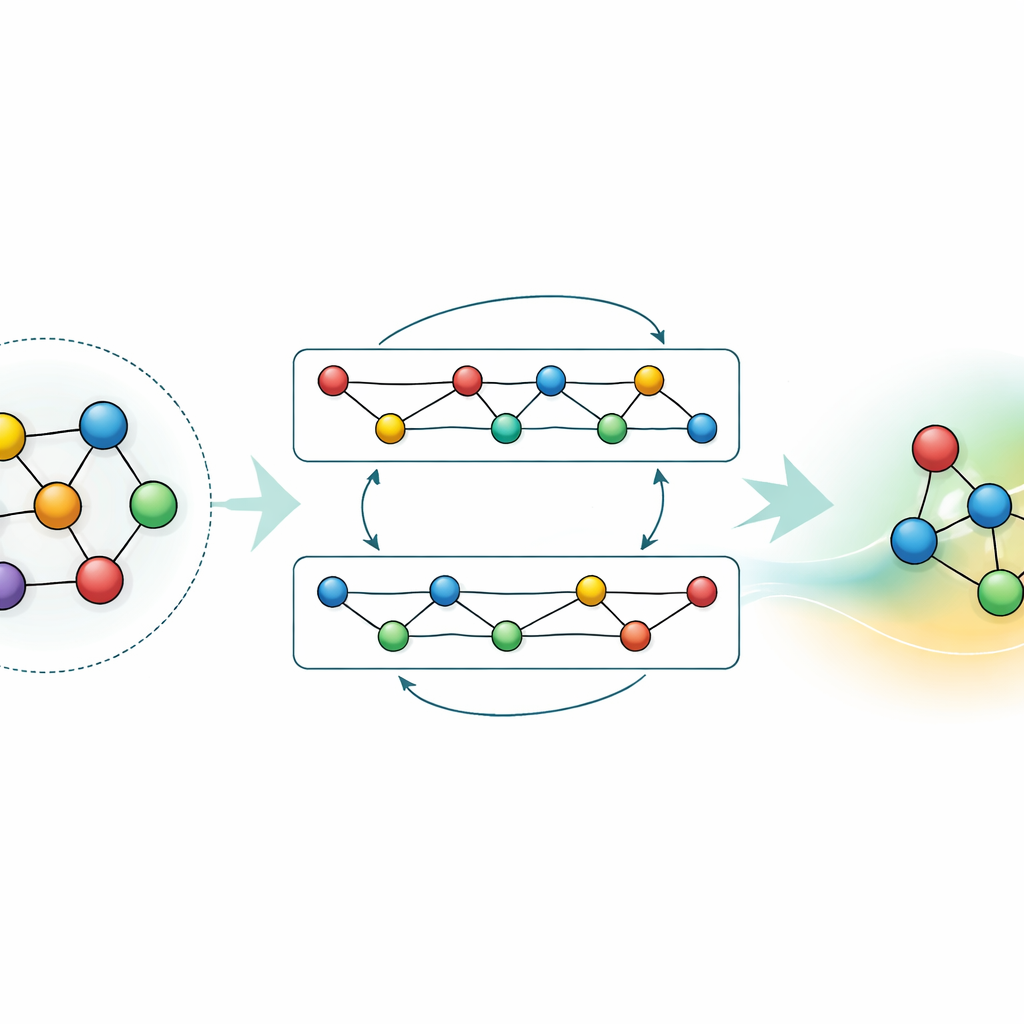
Adapting and Compressing the Knowledge
While a general-purpose model is powerful, many applications need either higher accuracy for a specific material or faster calculations on modest hardware. The authors demonstrate two strategies to achieve this without throwing away what GRACE has already learned. First, they fine-tune the foundational model on focused datasets, such as aluminum–lithium alloys or detailed hydrogen combustion pathways. For the alloys, even modest extra data significantly sharpens predictions, outperforming models trained from scratch with the same information. For combustion, naive fine-tuning would normally cause the model to “forget” what it knew about other materials; by carefully freezing parts of the network and only updating selected parameters, the authors limit this catastrophic forgetting while still gaining accuracy for the new chemistry. Second, they show how to distill the large model into a much simpler “student” that mimics the teacher on key systems. This distilled version runs around seventy times faster on a CPU yet preserves most of the accuracy, especially when trained on a mix of complex alloys and simpler reference structures labeled by the original GRACE.
What This Means for Future Materials Design
The work positions GRACE as a flexible foundation for the next generation of atomistic modeling. Instead of crafting a new potential for every material or property, researchers can start from a universal GRACE model and then fine-tune or distill it to their needs, saving vast amounts of computer time and expert labor. The benchmarks show that this approach does not merely match existing tools; it often surpasses them in both speed and reliability, particularly for demanding properties like thermal transport. For non-specialists, the key message is that a single, well-designed machine-learning model can now act as a broadly trusted “engine” for virtual experiments across much of the periodic table, accelerating the search for better batteries, catalysts, structural alloys, and energy materials.
Citation: Lysogorskiy, Y., Bochkarev, A. & Drautz, R. Graph atomic cluster expansion for foundational machine learning interatomic potentials. npj Comput Mater 12, 114 (2026). https://doi.org/10.1038/s41524-026-01979-1
Keywords: machine learning interatomic potentials, materials modeling, atomic simulations, foundational models, graph atomic cluster expansion