Clear Sky Science · en
Origin of the machine learning forces field errors across metal elements
Why some metals are harder for AI to understand
Machine-learning models are becoming powerful tools for simulating how atoms move, saving scientists huge amounts of computer time compared with traditional quantum calculations. You might expect that the simplest materials in nature—pure metals made of a single element—would be the easiest for these models to learn. This study shows that is not true: some metals remain stubbornly difficult to describe, and the authors uncover a physical reason why.
Building a big, clean map of metallic behavior
To look systematically at this problem, the researchers created a new dataset called Metal-43, based on demanding quantum-mechanical calculations. It covers 43 different metallic elements, from lightweight lithium to heavy tungsten, all treated with consistent computational settings. For each metal, they simulated thousands of atomic structures at several temperatures, recording the energy and forces on every atom. This carefully controlled “playground” lets them test machine-learning force fields—AI models that predict atomic forces—under fair, comparable conditions across many metals. 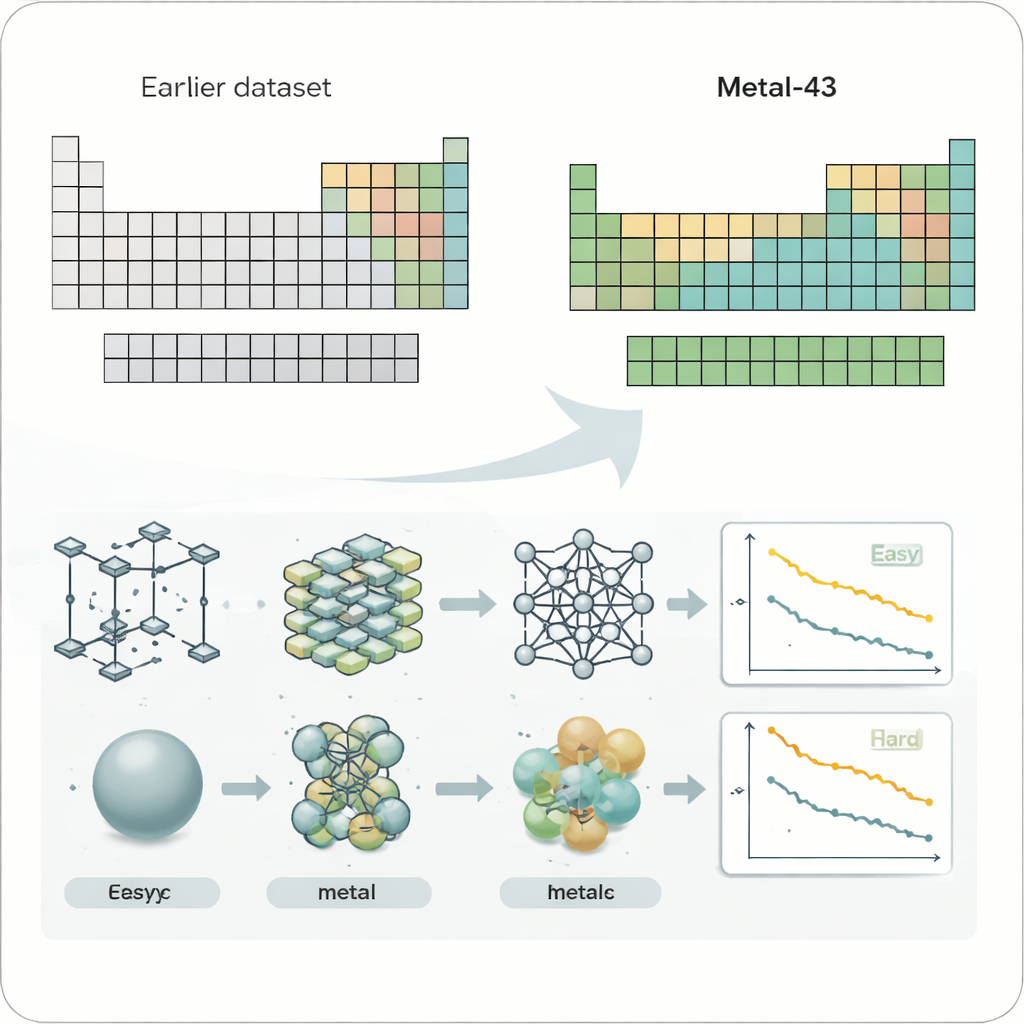
How model errors line up with the periodic table
Four widely used machine-learning force-field models were examined, including both compact models trained separately for each element and large, general-purpose models trained on many systems at once. When the authors charted the prediction errors on a periodic-table layout, a striking pattern appeared. Soft, more weakly bonded metals such as alkali and alkaline-earth elements tended to be easier for every model, while early transition metals in the middle of the table—those often used in high-performance alloys and catalysts—consistently produced much larger errors. This trend held even when the raw errors were rescaled to account for the overall strength of the atomic forces, showing that the difficulty is not just a matter of stronger bonds but something more fundamental.
Hidden complexity in the metal’s “traffic map” of electrons
The key insight of the work is to connect these model errors to the shape of each metal’s Fermi surface, which is a kind of three-dimensional “traffic map” of where electrons can move at the energies that matter most. In easy-to-fit metals, this surface is smooth and close to spherical. In hard-to-fit early transition metals, it becomes jagged and pocketed, reflecting complicated electron behavior tied to partially filled d orbitals. When atoms are jiggled or slightly displaced, these intricate Fermi surfaces change in an uneven, sometimes abrupt way, which in turn makes the overall energy landscape rough and complex. The authors show that simple numerical measures of how rapidly certain electron energy sums wiggle under small perturbations line up strongly with how large the machine-learning errors are, especially for those problematic transition metals. 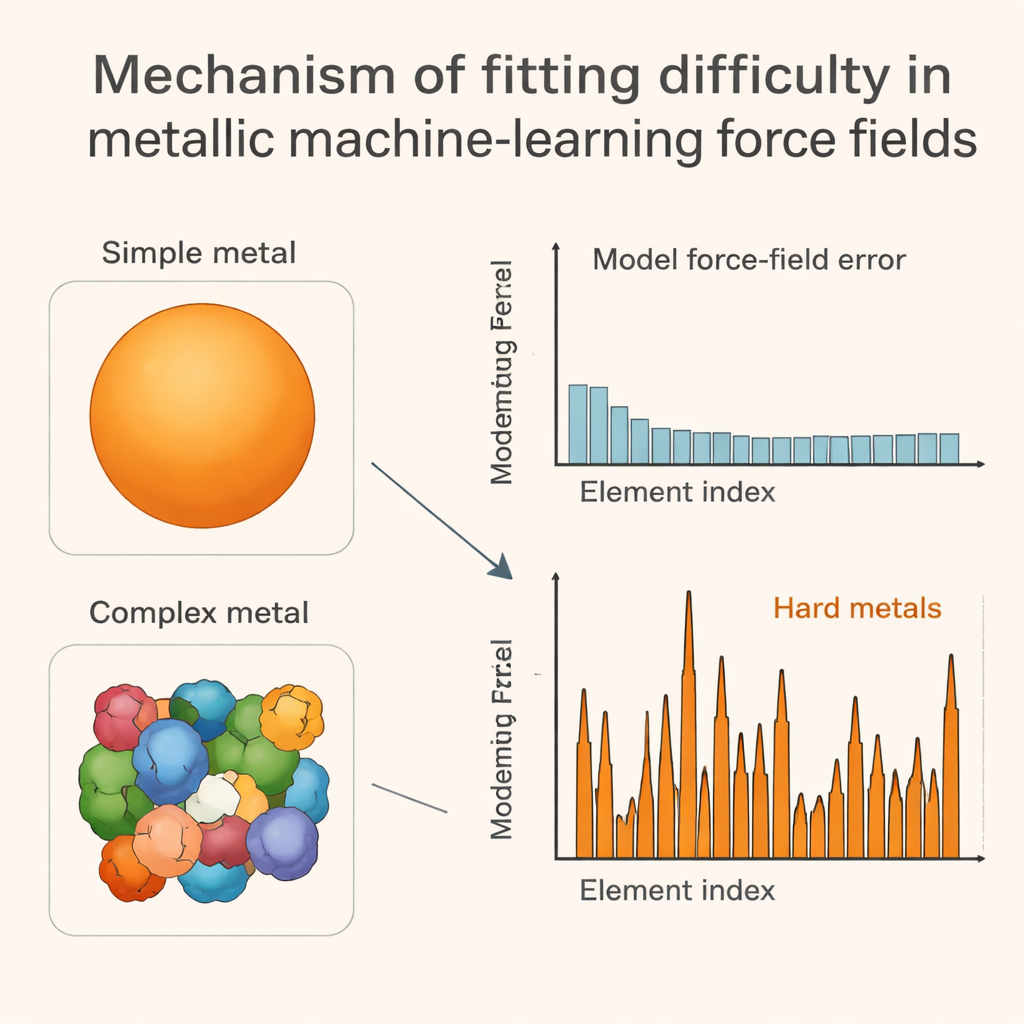
Limits of current AI models, even on idealized data
To separate the difficulty of the metals themselves from the limits of current AI approaches, the team also generated artificial datasets using traditional, hand-crafted models of atomic forces. Some of these older models depend mainly on distances between atoms, while others include strong angle dependence that mimics more directional bonding. Machine-learning force fields could reproduce the distance-based models almost perfectly, but their errors grew sharply when angular effects were important—particularly for metals already known to be hard. This comparison shows that the challenge lies not only in the underlying physics of the metals but also in the representational power of today’s machine-learning architectures, which still struggle with strongly angle-dependent, many-body interactions.
What this means for future simulations
For non-specialists, the main conclusion is that there is a clear, physically grounded reason why some metals are much harder for AI to model than others: the complexity of how their electrons move at the Fermi level makes the energy landscape rough and intricate. The Metal-43 dataset and the simple electronic-structure indicators proposed here give researchers a way to anticipate which materials will be troublesome, to benchmark new models fairly, and to design improved force fields that better capture directional bonding. In the long run, these insights should help make AI-based simulations more reliable for designing advanced alloys, catalysts, and other metal-based technologies.
Citation: Geng, X., Zhang, W., Wang, LW. et al. Origin of the machine learning forces field errors across metal elements. npj Comput Mater 12, 102 (2026). https://doi.org/10.1038/s41524-026-01977-3
Keywords: machine learning force fields, metallic materials, Fermi surface, interatomic potentials, density functional theory