Clear Sky Science · en
Self-optimizing machine learning potential assisted automated workflow for highly efficient complex systems material design
Smarter Searches for New Materials
Designing new materials is a bit like looking for a needle in a near-infinite haystack. From better batteries and faster computers to more efficient lasers and potential room-temperature superconductors, many future technologies depend on discovering just the right atomic arrangements. This paper presents a way to let artificial intelligence do most of that searching automatically, dramatically cutting the time and cost needed to find promising new compounds.
Why the Materials Puzzle Is So Hard
The properties of a solid—how well it conducts electricity, how strong it is, how it responds to light—are determined by how its atoms are arranged in three-dimensional patterns called crystal structures. In theory, one can use quantum mechanics to calculate which arrangements are stable and what their properties will be. In practice, these quantum calculations are so demanding that only a tiny fraction of all possible materials can be checked. The challenge grows rapidly when more than two chemical elements are involved, because the number of combinations and atomic arrangements explodes, making a blind search impossible.
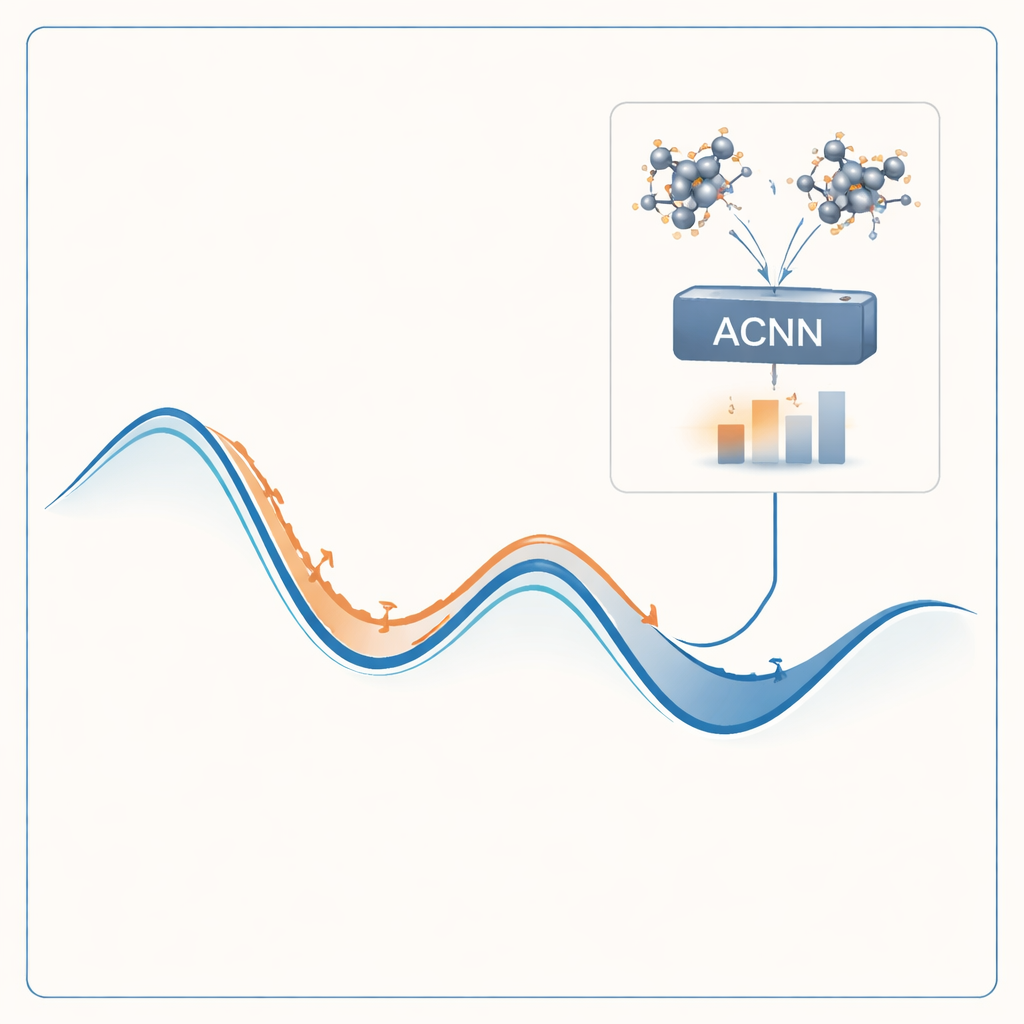
Letting a Learning Model Stand In for Quantum Physics
To tackle this problem, the authors build a machine-learning model that can imitate the results of expensive quantum calculations at a tiny fraction of the cost. Their model, called an attention-coupled neural network (ACNN), learns how the energy of a material depends on the positions and types of its atoms. Once trained, it can very quickly estimate whether a proposed crystal structure is likely to be stable or not, and what forces act on each atom. Crucially, the model is designed to respect basic physical requirements, such as the fact that shifting or rotating the entire crystal should not change its total energy.
A Self-Improving Materials Discovery Loop
Rather than training the model once and hoping it works everywhere, the authors wrap it in a self-optimizing loop. The process begins with a small set of random crystal structures, which are evaluated with full quantum-mechanical calculations and used to train an initial ACNN. This model is then used to relax millions of trial structures, quickly finding local energy minima—candidate stable or nearly stable phases. The workflow automatically flags two especially valuable kinds of structures: those that look very stable and those that appear unphysical or suspicious. Only these selected cases are sent back to the expensive quantum solver, and the new results are fed into the model for retraining. Over many rounds, the model becomes steadily more accurate in the regions of structure space that matter most.
Putting the Method to the Test
The team demonstrates their approach on two demanding systems. The first is a high-pressure mixture of magnesium, calcium, and hydrogen, a family of compounds of great interest for high-temperature superconductivity. By exploring nearly six million trial structures, their workflow uncovers a new stable phase, MgCa₃H₂₃, and several closely related hydrogen-rich “cage” structures. Calculations suggest that some of these could superconduct at temperatures above the boiling point of liquid nitrogen under extreme pressure. The second test targets a four-element system containing beryllium, phosphorus, nitrogen, and oxygen, chosen for its potential to host crystals that efficiently convert laser light into deep ultraviolet wavelengths. Here the method relaxes more than nine million structures and identifies three thermodynamically stable phases with very wide band gaps and promising optical characteristics.
From Brute Force to Guided Discovery
Across both examples, the automated workflow achieves speedups of about ten thousand times compared with using quantum calculations alone, while still reliably pinpointing structures worth closer study. For a non-specialist, the key message is that a large part of materials discovery can now be handled by a learning system that teaches itself where it is uncertain and asks for targeted, high-precision calculations only when needed. This kind of self-correcting, AI-assisted search opens the door to exploring far more complex mixtures of elements than was previously feasible, increasing the chances of uncovering new superconductors, optical crystals, and other functional materials that underpin next-generation technologies.
Citation: Li, J., Feng, J., Luo, J. et al. Self-optimizing machine learning potential assisted automated workflow for highly efficient complex systems material design. npj Comput Mater 12, 101 (2026). https://doi.org/10.1038/s41524-026-01971-9
Keywords: materials discovery, machine learning potentials, crystal structure prediction, superconducting hydrides, nonlinear optical crystals