Clear Sky Science · en
Host-derived interleukin-1α drives tumor immunosuppression by reprogramming tumor-associated myeloid cells
Why this research matters for cancer patients
Immunotherapy has transformed treatment for some cancers, but many breast tumors still evade the immune system. This paper explores a lesser-known culprit: a signaling molecule called interleukin‑1 alpha (IL‑1α) made not by the cancer cells themselves, but by normal cells in the body. The authors show that host‑derived IL‑1α can quietly turn immune cells into allies of the tumor, and that blocking this signal in mice can cause breast tumors to shrink or disappear. Understanding this switch could help doctors design treatments that make immunotherapy more effective for breast cancer.
A hidden influencer in the tumor neighborhood
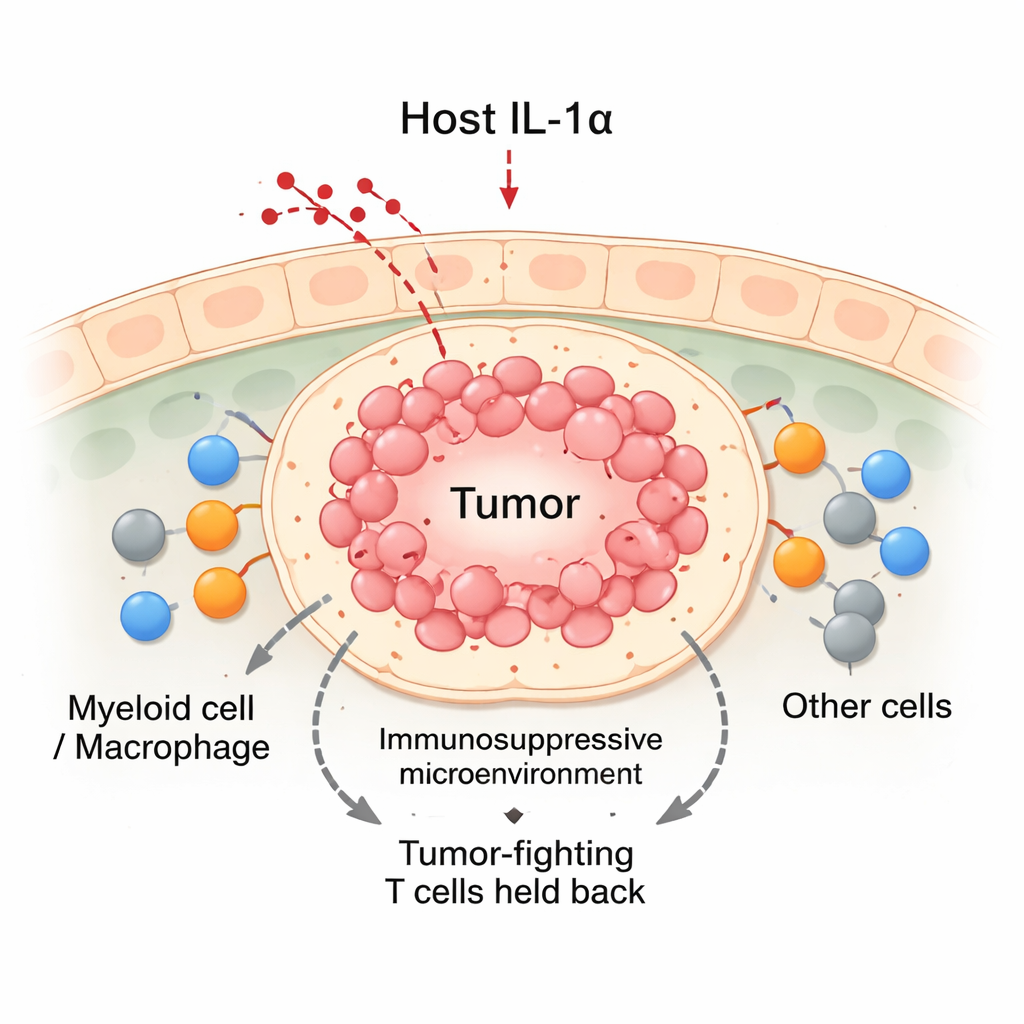
Cancers do not grow in isolation; they live in a busy “neighborhood” of immune cells, blood vessels, and connective tissue known as the tumor microenvironment. Among the most important residents are myeloid cells, especially macrophages, which can either attack tumors or protect them. Classic textbooks divide macrophages into M1 (tumor‑fighting) and M2 (tumor‑supporting), but real tumors contain many shades in between. The authors focused on IL‑1α, a molecule normally produced by non‑cancerous tissues, to ask whether this host signal steers incoming myeloid cells toward helpful or harmful roles inside breast tumors.
Switching off IL‑1α tips the balance against tumors
Using mouse models of breast cancer, the team compared normal animals with others genetically engineered to lack IL‑1α. When breast cancer cells were transplanted into the mammary glands, tumors in normal mice grew steadily. In contrast, tumors in IL‑1α‑deficient mice initially grew for about two weeks and then frequently regressed. These regressing tumors contained more immune cells overall, including a surge of CD8 “killer” T cells and myeloid cells. Even though IL‑1α‑deficient mice had fewer white blood cells circulating in their blood, they packed many more immune cells into the tumor site. Detailed analyses showed that the infiltrating CD8 T cells in IL‑1α‑deficient tumors were more active, producing higher levels of tumor‑killing molecules and showing fewer signs of exhaustion.
How IL‑1α reshapes tumor‑associated myeloid cells
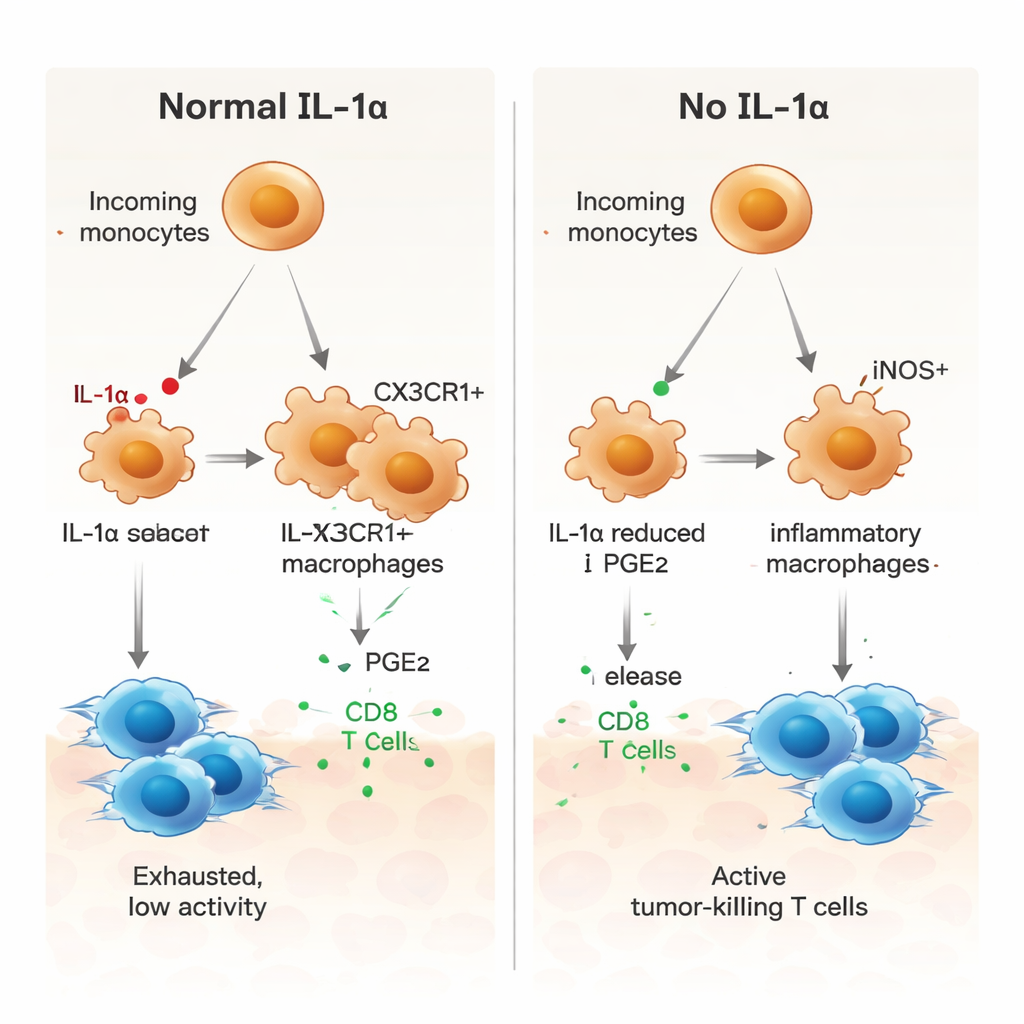
To understand what IL‑1α was doing at the cellular level, the researchers used single‑cell RNA sequencing to examine thousands of individual cells from tumors. They discovered that only a small subset of tumor‑associated macrophages—those marked by a receptor called CX3CR1—produced IL‑1α. In normal mice, many incoming monocytes matured into these CX3CR1‑positive macrophages, which were linked to immune‑suppressive behavior. In mice lacking IL‑1α, this route of maturation was disrupted. Instead, myeloid cells were more likely to become iNOS‑positive inflammatory macrophages, a profile associated with anti‑tumor activity. Functionally, myeloid cells from IL‑1α‑deficient tumors were less able to induce brakes such as PD‑1 and CTLA‑4 on T cells, and showed weaker capacity to dampen T‑cell proliferation.
A key role for a lipid messenger, PGE2
The authors then asked how IL‑1α drives myeloid cells toward a tumor‑supporting state. Communication analysis of the single‑cell data pointed to several signaling pathways, including one involving prostaglandin E2 (PGE2), a lipid mediator known to promote immune suppression. In tumors lacking IL‑1α, macrophages expressed lower levels of receptors for macrophage growth factor (M‑CSF), TGF‑β receptors, and a PGE2 receptor, and nearby stromal cells made less of the enzymes that produce PGE2. In laboratory cultures, bone‑marrow‑derived macrophages from IL‑1α‑deficient mice showed a more inflammatory profile, with higher iNOS and lower CX3CR1. Adding PGE2 reversed this shift and restored a more suppressive pattern, while blocking PGE2 in normal macrophages pushed them toward a pro‑inflammatory state. Macrophages without IL‑1α also stimulated tumor‑specific CD8 T‑cell proliferation more strongly, an effect blunted when PGE2 was added back.
Bridging mouse findings to human immunity
To see whether their mouse data reflect human biology, the team compared gene signatures from the different mouse macrophage subsets to a large panel of human myeloid cells driven toward various states in the lab. Macrophage clusters from tumors in normal mice resembled human cells pushed toward M2‑like, immune‑dampening states by molecules such as IL‑10, IL‑4, and glucocorticoids. By contrast, the same clusters from IL‑1α‑deficient tumors matched human cells in more inflammatory, less suppressive conditions. Notably, gene patterns linked to PGE2 signaling were enriched in IL‑1α‑dependent macrophages but absent when IL‑1α was missing, reinforcing the idea that IL‑1α and PGE2 work together to build an immunosuppressive niche.
What this could mean for future breast cancer therapy
In plain terms, this study suggests that a host‑derived molecule, IL‑1α, can quietly tell certain macrophages to protect the tumor and keep killer T cells in check, in part through PGE2 signaling. When IL‑1α is removed in mice, macrophages shift toward a more inflammatory, tumor‑attacking state, CD8 T cells become more active, and transplanted breast tumors are often rejected. Because current immunotherapies already aim to re‑energize exhausted T cells, combining them with approaches that block IL‑1α or its downstream PGE2 pathway could further tilt the tumor microenvironment in favor of the patient’s immune system. While more work is needed to test safety and effectiveness in humans, these findings highlight IL‑1α as a promising new target to make breast cancer more vulnerable to immune attack.
Citation: Keerthi Raja, M.R., Gupta, G., Atkinson, G. et al. Host-derived interleukin-1α drives tumor immunosuppression by reprogramming tumor-associated myeloid cells. npj Breast Cancer 12, 26 (2026). https://doi.org/10.1038/s41523-026-00890-8
Keywords: breast cancer immunotherapy, tumor microenvironment, macrophages, interleukin-1 alpha, prostaglandin E2