Clear Sky Science · en
Learning data-efficient coarse-grained molecular dynamics from forces and noise
Why shrinking molecules matters
Simulating the restless motion of every atom in a protein and its surrounding water is one of our best tools for understanding how life works at the molecular scale. But these all-atom simulations are so computationally demanding that following a protein as it folds, unfolds, or interacts with partners for biologically relevant times can take months on a supercomputer. This article introduces a new way to build fast, simplified models of proteins that still behave like their full atomic counterparts, while needing far less training data and computing power than before.
From every atom to a simpler picture
Traditional molecular dynamics tracks each atom and computes the forces between them at every tiny time step. To speed things up, scientists often use coarse-grained models, which group many atoms into a smaller number of “beads.” These reduced models run much faster but have historically struggled to match the accuracy of full atomistic simulations, especially for proteins with rich folding behavior. Recent work has turned to machine learning to automatically discover better coarse-grained force fields, but training these models has typically required millions of detailed snapshots, each labeled with the forces on every atom—an enormous data and computing burden.
Blending physical forces with informative noise
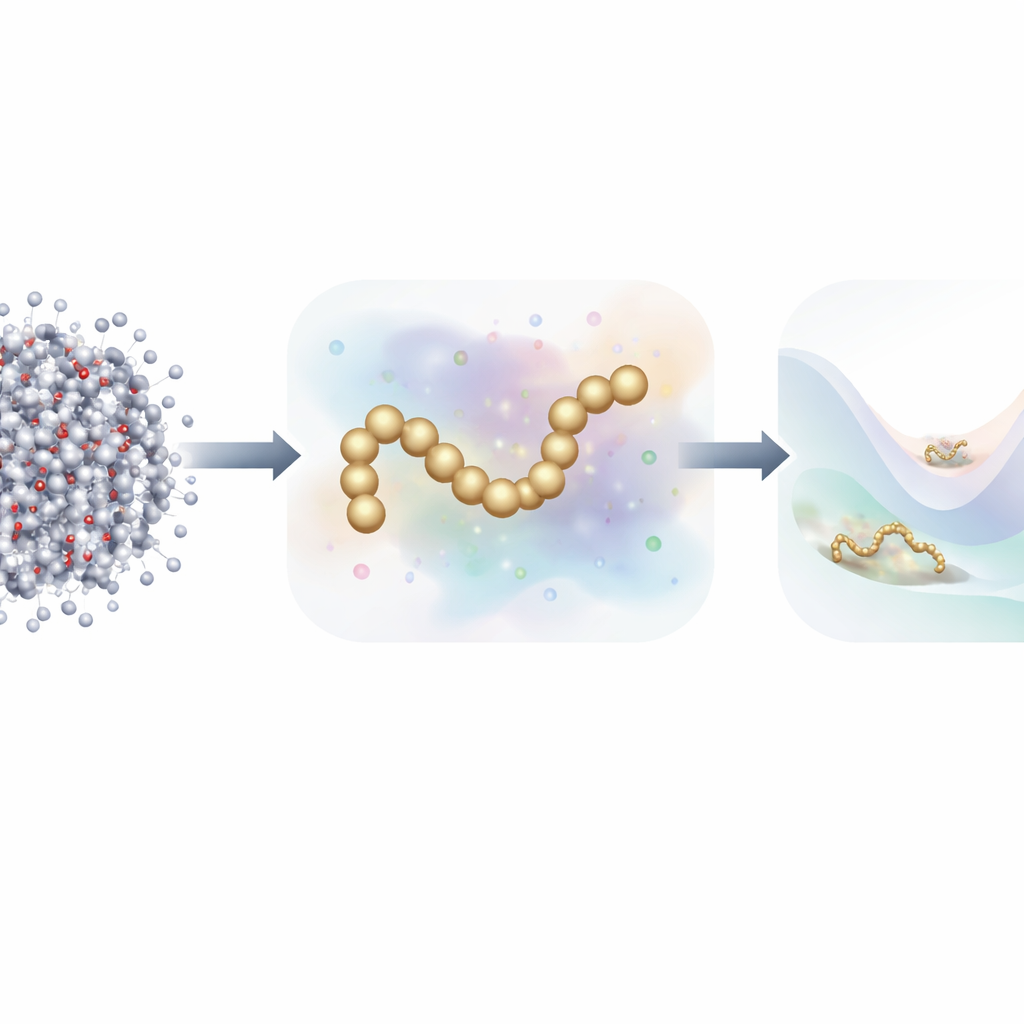
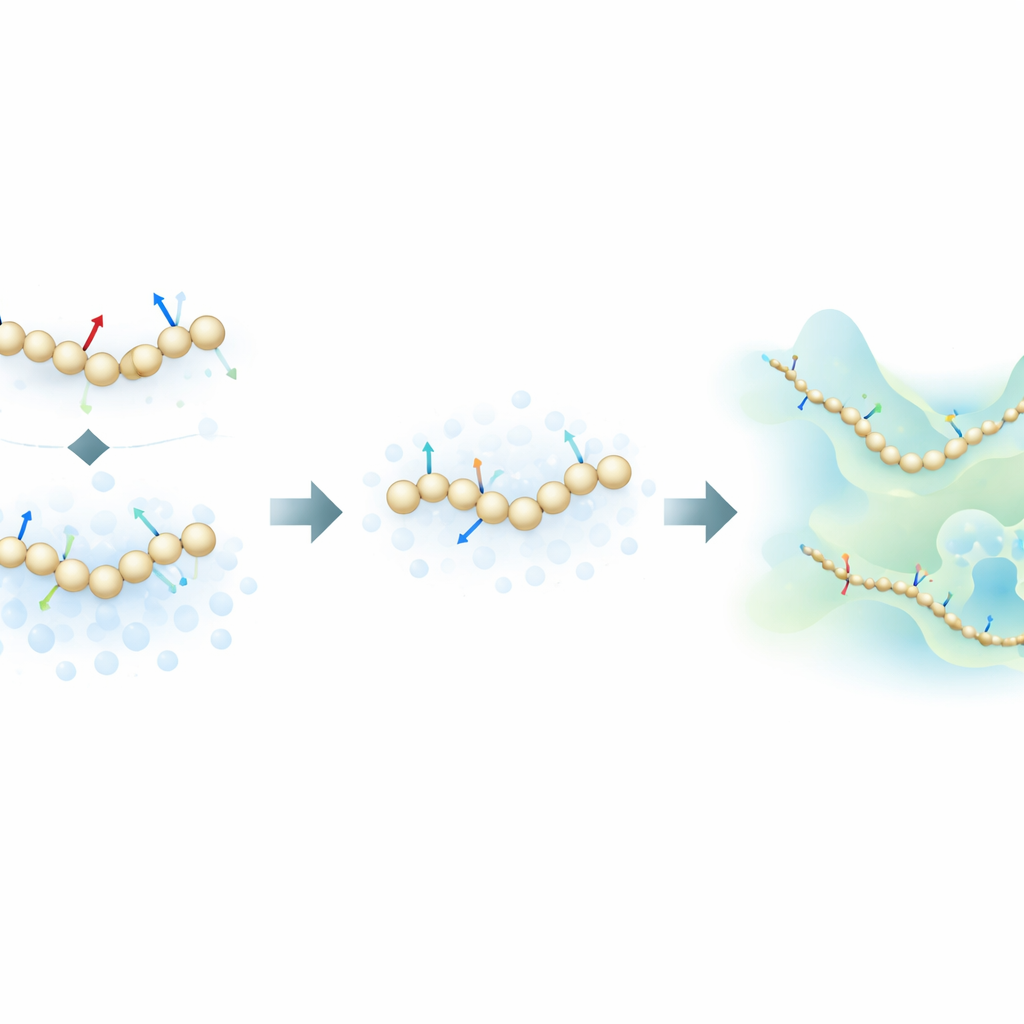
The authors propose a fresh training strategy that takes inspiration from generative diffusion models—the same class of algorithms behind many modern AI image generators. Instead of learning only from the physical forces calculated in atomistic simulations, their method also learns from how molecular structures are distributed in space by deliberately adding controlled noise to coarse-grained configurations. In this framework, noise is not just a nuisance to be removed; it becomes an extra source of information. By mathematically unifying the traditional “force matching” approach with denoising techniques from diffusion models, the method can infer the underlying energy landscape of a protein using far fewer labeled examples.
Teaching simple models to mimic complex proteins
To test their idea, the researchers trained neural-network coarse-grained models for several proteins of increasing complexity: the small miniproteins Chignolin and Trp-Cage, the somewhat larger NTL9, and the 76-residue protein Ubiquitin. They compared three training modes: using only atomistic forces, using only noise-based information, and combining both. For the smaller proteins, they showed that the new combined approach can reproduce the key features of the folding landscape—such as the relative stability of folded and unfolded states and the presence of intermediates—using up to one hundred times less training data than standard force-matching methods. Surprisingly, in data-scarce regimes, even models trained only with noise-based information often matched or exceeded the accuracy of force-only training.
Reaching larger and tougher protein systems
Ubiquitin is a more demanding test: capturing its folding and unfolding at realistic temperatures has historically required specialized hardware and extremely long atomistic runs. Here, the authors train coarse-grained models using a modest dataset consisting of short equilibrium simulations around the folded state plus non-equilibrium “pulled” simulations that forcibly stretch the protein. Despite this biased training set and the lack of a perfect atomistic reference at the same conditions, the model trained with both forces and noise recovers a realistic picture in which folded and unfolded states coexist, with the folded state favored in stability. In contrast, a model trained only on forces fails to stabilize the folded state at all, while a noise-only model prefers unfolded structures. Notably, none of the coarse-grained models simply memorizes the extreme stretched shapes from the training data, indicating that the learned energy landscape is physically meaningful and not just an imprint of the input trajectories.
What this means for future simulations
By turning noise into a training signal and merging it with physical forces, this work shows that accurate coarse-grained models of proteins can be built from far smaller and less ideal datasets than previously thought. In practice, that means researchers may no longer need millisecond-long atomistic simulations on specialized supercomputers before they can explore a biomolecule’s behavior with machine-learned coarse-grained dynamics. Instead, more modest simulations on widely available hardware could be enough to train powerful reduced models that capture key folding pathways and thermodynamic balances. While questions remain about how best to choose and interpret the added noise and how the method will fare on even larger, more complex biomolecular assemblies, this approach substantially lowers the barrier to using data-driven coarse-grained simulations as a routine tool in molecular science.
Citation: Durumeric, A.E.P., Chen, Y., Pasos-Trejo, A.S. et al. Learning data-efficient coarse-grained molecular dynamics from forces and noise. Nat Commun 17, 2493 (2026). https://doi.org/10.1038/s41467-026-70818-0
Keywords: coarse-grained molecular dynamics, machine learning force fields, protein folding simulations, diffusion models in chemistry, data-efficient simulation