Clear Sky Science · en
A pathogenic Tau mutation drives autophagy-lysosome dysfunction that limits Tau degradation in a model of frontotemporal dementia
When the Brain’s Cleanup Crews Fall Behind
Why do some people develop devastating memory and behavior problems decades before old age? This study tackles that question by zooming in on a single brain protein, Tau, and the tiny cellular "recycling centers" that normally keep it in check. By watching living human nerve cells under super‑sharp microscopes, the researchers show how a disease‑causing Tau mutation clogs the cell’s waste‑disposal system and how revving up that system with a small molecule can help clear the mess. Their findings may point toward new treatment strategies for certain forms of dementia.
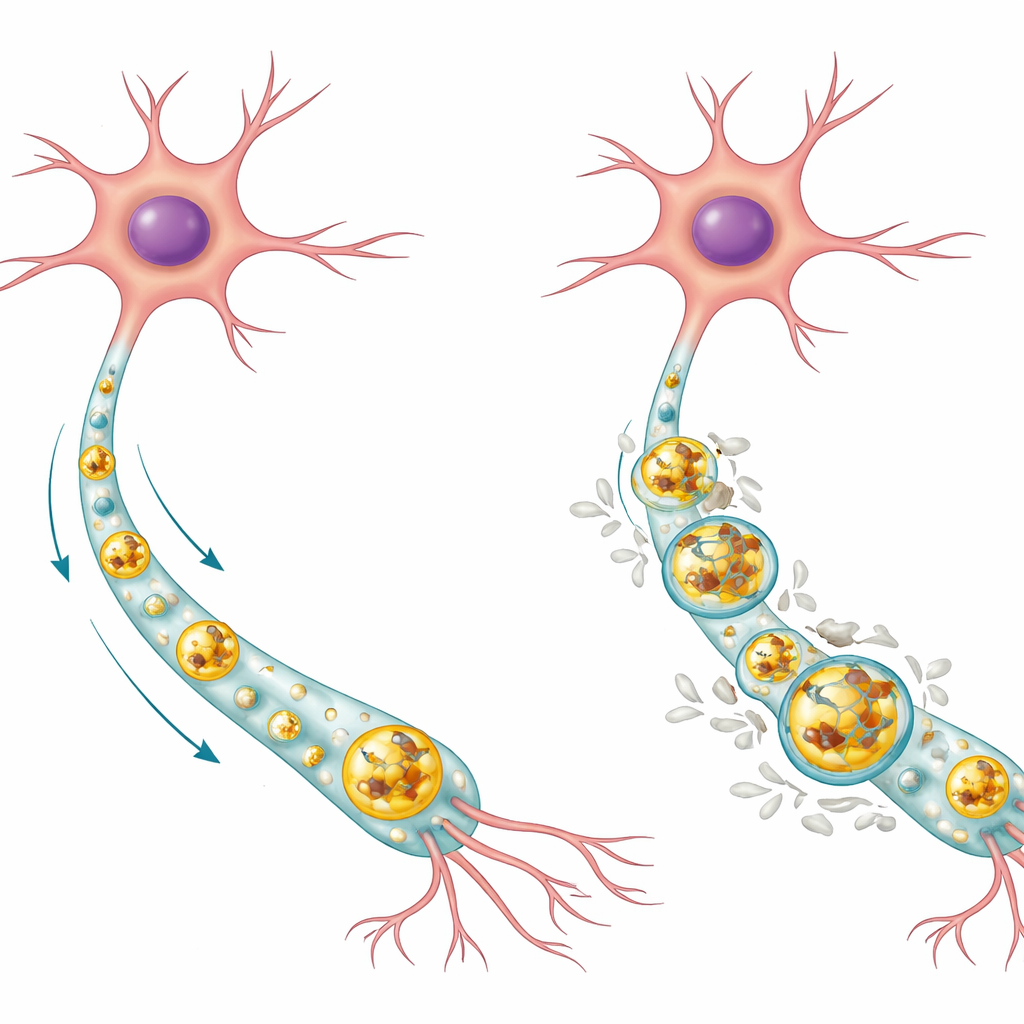
How Brain Cells Usually Take Out the Trash
Neurons are long‑lived cells that cannot simply divide to dilute damaged material, so they rely heavily on internal cleanup systems. One key route is the autophagy‑lysosome pathway. In this process, unwanted proteins and worn‑out parts are wrapped in membrane sacs called autophagosomes, which then fuse with enzyme‑filled compartments known as lysosomes, where the cargo is broken down and recycled. In healthy human neurons, the authors found that normal Tau protein tends to accumulate inside the acidic center of lysosomes, where it can be degraded, while the phosphorylated form of Tau (a chemical modification linked to disease) sits more on the lysosome’s outer membrane. Most lysosomes in healthy cells were free of Tau altogether, suggesting this system usually keeps Tau levels low and well controlled.
What Goes Wrong in a Genetic Form of Dementia
The team focused on a mutation in the MAPT gene, called p.R406W, which causes an inherited form of frontotemporal dementia and can mimic Alzheimer’s‑like memory loss. Using stem‑cell technology, they reprogrammed patient skin cells into induced pluripotent stem cells and then into large numbers of human neurons that either carried the mutation or had been gene‑edited back to normal. In the mutant neurons, total Tau and phosphorylated Tau were markedly higher, not because the cells produced more Tau, but because they cleared it less efficiently. Super‑resolution imaging revealed that nearly all lysosomes in the mutant cells were packed with Tau and especially with phosphorylated Tau coating the lysosome membrane. This build‑up signaled that the cell’s main protein disposal route was jammed.
Clogged Recycling Centers and Sluggish Traffic
Looking more closely at the recycling machinery, the researchers saw that lysosomes in mutant neurons were more numerous, larger, and tended to sit farther away from the cell body. Live imaging with fluorescent dyes showed that these lysosomes moved more slowly and traveled shorter distances along the nerve fibers, even though the underlying microtubule tracks looked normal. The mutant neurons also contained more autophagosomes, more of the cargo‑adaptor protein p62, and extra lipid droplets—signs that material was being tagged for disposal but not fully broken down. Using a pH‑sensitive reporter, they found that autophagosomes in mutant cells often failed to fuse properly with lysosomes, leading to a pileup of "half‑finished" recycling vesicles and broad defects in cellular cleanup, not just for Tau but for other cargo as well.
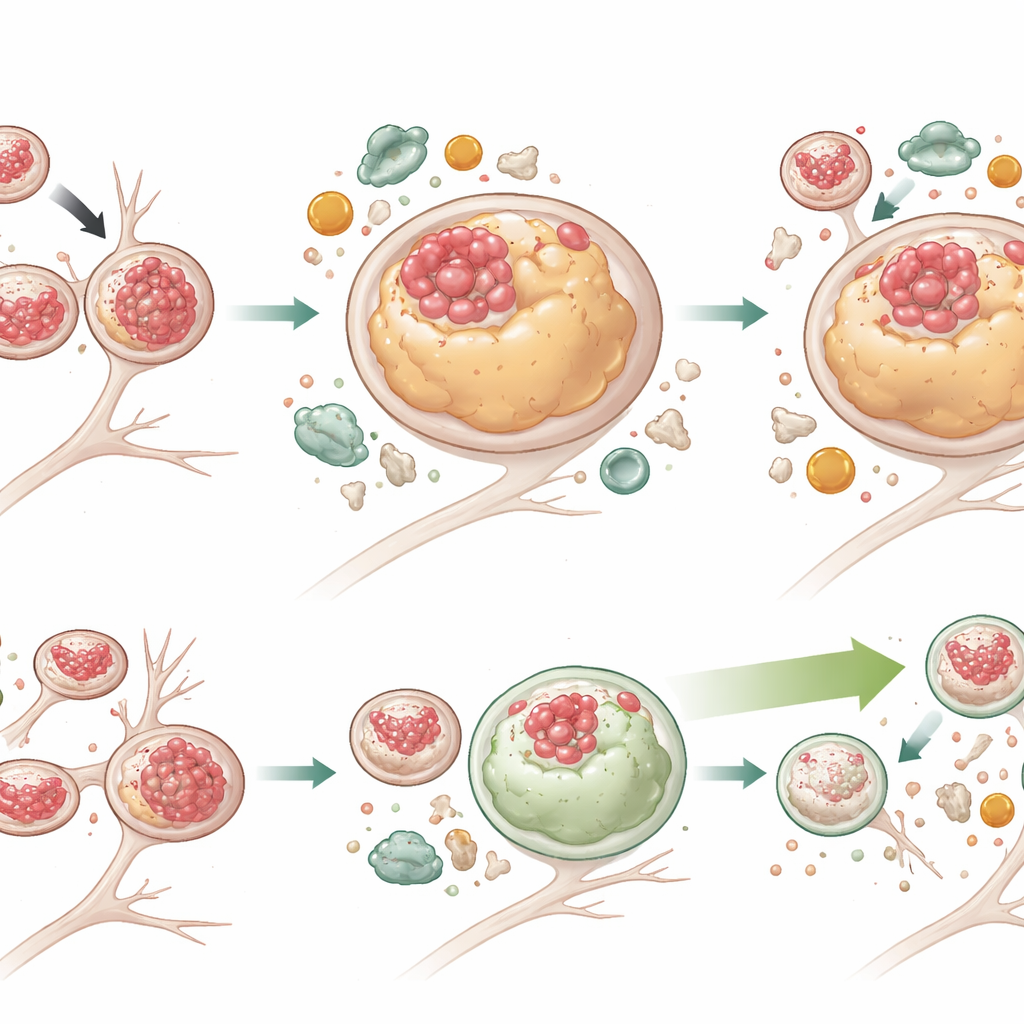
Boosting Cellular Cleanup Without Fixing the Traffic Jam
To test whether enhancing autophagy could overcome these problems, the team treated neurons with G2‑567, a small molecule previously shown to stimulate the autophagy‑lysosome system. After two weeks of treatment, mutant neurons had substantially lower levels of both total Tau and phosphorylated Tau, and many more lysosomes were once again free of Tau. Lysosomes also shrank back toward normal size. Markers of active autophagy increased, while p62—an indicator of stalled degradation—fell in mutant cells, demonstrating more effective cargo breakdown. Interestingly, G2‑567 did not correct all defects: lysosomes in mutant neurons still tended to sit farther from the cell body and move sluggishly, and an adaptor protein (JIP3) linked to lysosome transport remained elevated. This suggests that the movement and degradative functions of lysosomes can be partly uncoupled, and that improving breakdown alone may be enough to reduce toxic Tau buildup.
What This Means for Future Dementia Treatments
For a non‑specialist, the key takeaway is that in this genetic model of frontotemporal dementia, the problem is not simply that Tau becomes abnormal; it is that the neuron’s recycling system cannot keep up. The p.R406W Tau mutation directly disrupts several steps of the autophagy‑lysosome pathway, causing Tau—especially its phosphorylated form—to accumulate on and inside lysosomes, along with other undegraded material. By pharmacologically nudging the cell’s cleanup machinery to work harder, the researchers were able to lower Tau levels and normalize lysosome size, even though transport defects persisted. These findings strengthen the idea that drugs designed to safely boost autophagy and lysosomal function could help restore protein balance in tau‑related dementias and perhaps in more common conditions like Alzheimer’s disease.
Citation: Mirfakhar, F.S., Marsh, J.A., Sato, C. et al. A pathogenic Tau mutation drives autophagy-lysosome dysfunction that limits Tau degradation in a model of frontotemporal dementia. Nat Commun 17, 2699 (2026). https://doi.org/10.1038/s41467-026-70473-5
Keywords: tau protein, autophagy, lysosome dysfunction, frontotemporal dementia, neurodegeneration