Clear Sky Science · en
DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning
Seeing Proteins in New Detail
Proteins are the tiny machines that keep our cells alive, but fully reading their building blocks is still surprisingly hard. This paper introduces DiNovo, a new software system that helps scientists "read" protein fragments much more completely and reliably than before. By combining a clever biochemical trick with modern artificial intelligence, it promises to uncover hidden proteins, disease markers, and even immune targets that traditional methods often miss.
Why Reading Protein Fragments Is So Tough
Most protein analysis today relies on chopping proteins into smaller pieces, called peptides, and then weighing their fragments in a mass spectrometer. From these weights, computers try to reconstruct the original peptide sequence, like solving a crossword from partial clues. Existing methods usually assume that peptides come from known protein databases, which works well for familiar proteins but struggles with new or unexpected ones. So-called de novo sequencing avoids this limitation by trying to read peptides directly from the data, but it often falls short because some fragments are missing and some peptides are never cleanly cut in the first place.
Using Mirror Enzymes to Fill in the Gaps
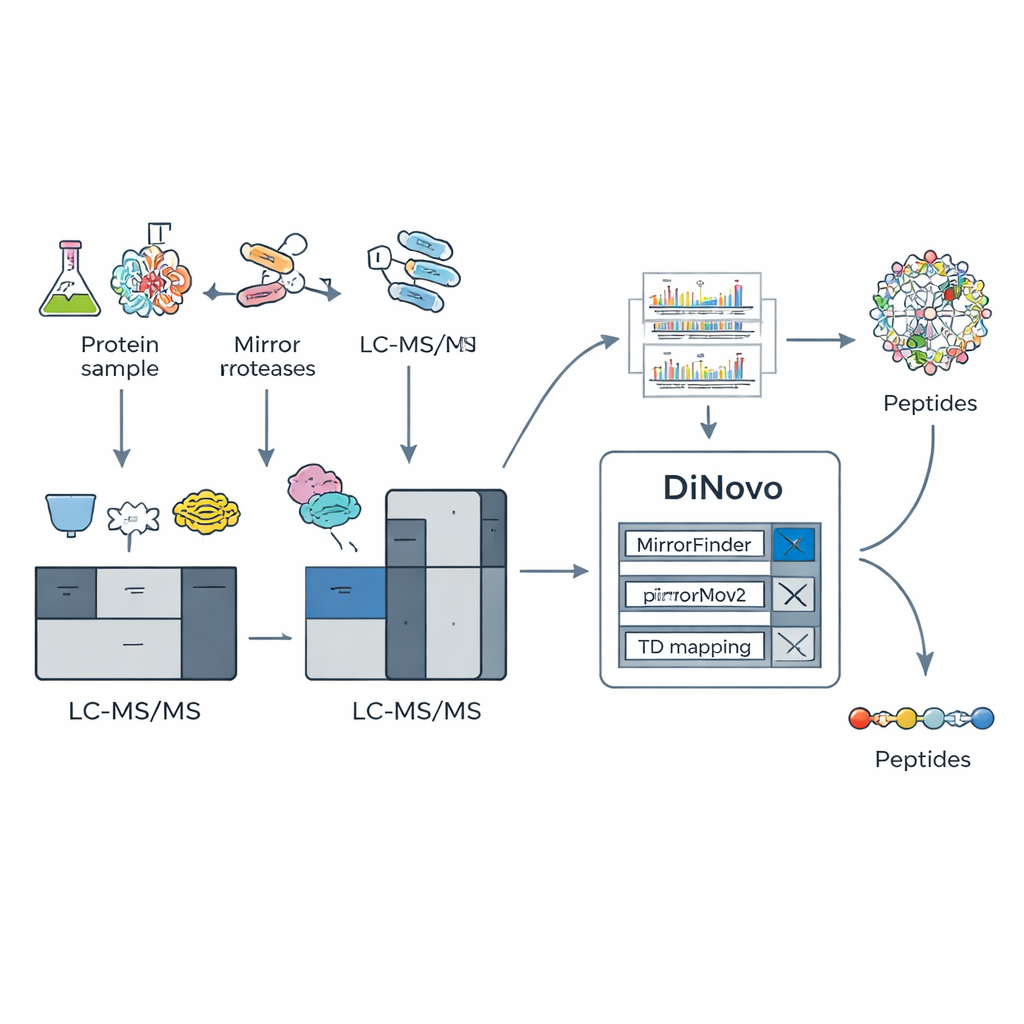
The key idea behind DiNovo is to use pairs of “mirror proteases” – pairs of cutting enzymes that slice proteins on opposite sides of the same type of amino acid. For example, one enzyme cuts just before a lysine, while its partner cuts just after that lysine. This produces two related peptides that share the same inner segment but have different ends. When these "mirror" peptides are analyzed, their mass spectra contain complementary fragment patterns: what is missing in one spectrum often appears in the other. The authors show that combining such mirror pairs can push fragment coverage close to complete, with about 98% of possible cuts supported by real experimental signals, far higher than what is seen when using a single enzyme alone.
A Smart Software Pipeline Built for Mirror Data
To exploit this biochemical trick, the team built DiNovo as an end-to-end software workflow. First, proteins from bacteria and yeast are digested with two mirror pairs of enzymes, and the resulting peptides are analyzed by high-resolution mass spectrometry. DiNovo then uses a module called MirrorFinder to automatically recognize which pairs of spectra come from mirror peptides, doing this directly from signal patterns rather than from any prior sequence guesses. Next, its main de novo engine, MirrorNovo, uses deep learning to interpret those paired spectra, while a backup graph-based engine, pNovoM2, offers a faster CPU-only option. Together, these tools translate peaks into amino acid sequences and also examine the individual spectra that did not form obvious pairs, squeezing out as much information as possible.
Measuring Trust Without Relying on Old Databases
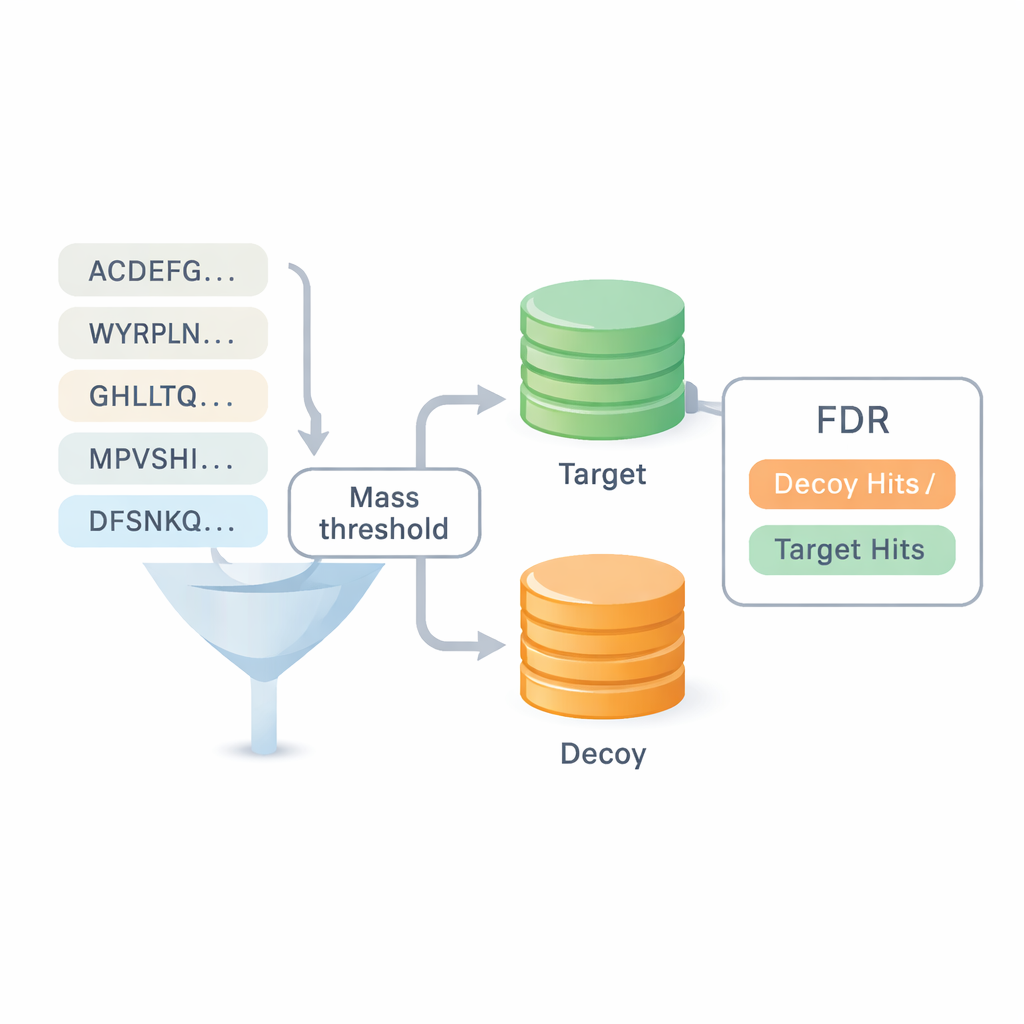
One of the biggest questions in de novo sequencing is how much to trust the results. Most existing benchmarks recycle answers from database search, which blurs the line between the two approaches and can hide errors. DiNovo introduces a different quality-check method called target-decoy mapping. Here, the newly read peptides are mapped to a combined collection of real (target) and artificial, scrambled (decoy) protein sequences. By comparing how often peptides land in the real set versus the scrambled set, the software can estimate an error rate, or false discovery rate, without leaning on previous identifications. This makes it possible to compare DiNovo directly to standard database search programs under the same error controls.
What DiNovo Delivers in Practice
In tests on bacterial, yeast, and antibody samples, DiNovo consistently read many more peptides and amino acids than well-known de novo tools that use only a single enzyme. Using two mirror pairs, it produced 2–3 times more high-confidence amino acids than a classic trypsin-only setup and identified more proteins at similar error levels. When compared directly to three leading database search engines, DiNovo found similar numbers of amino acids and proteins, and most of its sequences agreed with those from the search engines on the same spectra. The authors argue that this level of coverage and agreement means de novo sequencing, long treated as a backup method, can now stand beside database search as a serious, and in some cases superior, option.
Big Picture: Toward Complete, Unbiased Protein Reading
For a non-specialist, the takeaway is that DiNovo makes it far easier to read protein pieces accurately without being limited to what is already in reference databases. By doubling or tripling the amount of well-supported sequence information and providing its own built-in error checks, this approach opens the door to discovering unfamiliar proteins, tracking subtle variations, and exploring complex mixtures where many components are still unknown. In short, by pairing mirror enzymes with deep learning and careful statistics, DiNovo helps turn noisy spectral traces into a clearer, more trustworthy picture of the proteins that underlie health and disease.
Citation: Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
Keywords: proteomics, de novo peptide sequencing, mass spectrometry, deep learning, mirror proteases