Clear Sky Science · en
Single-cell multiomics uncovers an endothelial mechanosensitive PIEZO1-IL-33 axis driving pulmonary fibrosis
Why Stiff Lungs Matter
Pulmonary fibrosis is a devastating lung condition in which once‑springy air sacs slowly turn into stiff scar tissue, making every breath feel like hard work. Doctors can currently only slow the disease, not stop or reverse it. This study asks a deceptively simple question with big implications: how do the cells lining lung blood vessels sense that the tissue around them has become abnormally stiff, and how does that sensation get turned into more scarring? By tracing this chain of events down to single cells and individual genes, the researchers uncover a mechanical “switch” in blood vessels that may be druggable.
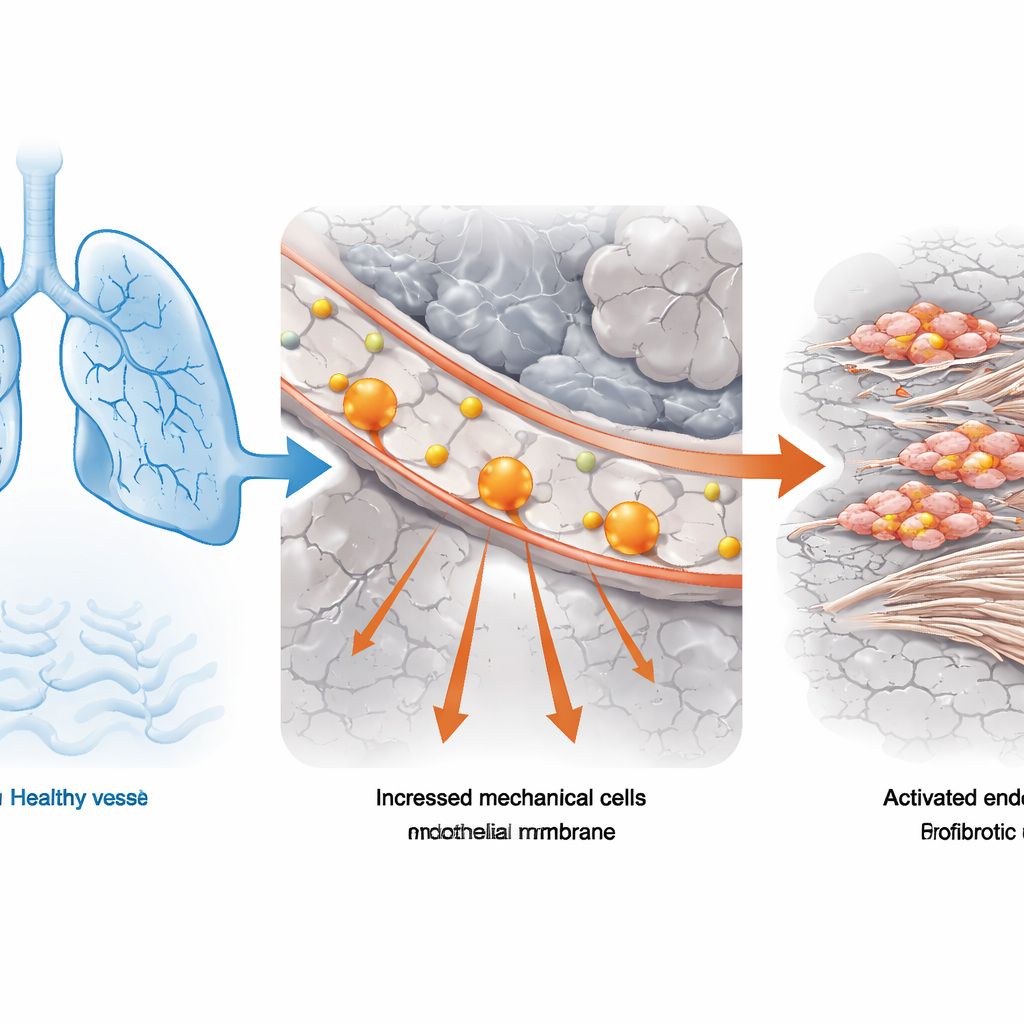
From Lung Function Tests to Single Cells
The team began with lung tissue from people with idiopathic pulmonary fibrosis, the most common form of the disease, and from donors with normal lungs. They combined standard lung function measurements (how much air a person can forcefully exhale) with powerful single‑cell RNA sequencing, which reads out which genes are active in thousands of individual cells. Using a computational tool that links bulk clinical data to single‑cell profiles, they pinpointed which cell types were most closely associated with severely impaired lung function. Vascular endothelial cells—the cells that form the inner lining of blood vessels—stood out as especially enriched in patients whose lung capacity was less than half of normal. In these endothelial cells, gene programs related to “mechanical stress” were consistently cranked up, suggesting that abnormal physical forces were part of the problem.
Mechanical Stress as a Hidden Driver
To test whether this link between stress‑sensing and disease was general, the researchers turned to two mouse models of lung scarring: one triggered by the chemotherapy drug bleomycin, the other by long‑term silica dust exposure, an occupational hazard. Using single‑cell methods on these experimental lungs, they again found that endothelial cells carried clear signatures of heightened mechanical stress. In both models, as the surrounding lung tissue thickened and stiffened, the blood‑vessel lining appeared to switch into a maladaptive state. This convergence across human samples and animal models strengthened the idea that distorted physical forces in the lung, rather than just inflammation or immune activity, are central to how fibrosis develops and progresses.
A Pressure Sensor with a Critical Role
Digging deeper, the team searched for specific “mechanosensors”—proteins that convert physical stretch into biochemical signals—that were upregulated in stressed endothelial cells. One channel protein, called PIEZO1, repeatedly emerged as a prime suspect. In both mice and humans with fibrosis, PIEZO1 levels were markedly higher in vascular endothelial cells than in healthy controls. When the researchers engineered mice in which PIEZO1 was deleted only in the endothelium, these animals were much more resistant to bleomycin‑induced lung scarring: they had less collagen buildup, fewer activated scar‑forming cells, and lower levels of a chemical marker of fibrosis. Pharmacologically blocking PIEZO1 with a peptide inhibitor also eased scarring, whereas activating it made fibrosis worse—unless the receptor was missing from endothelial cells. Together, these experiments showed that PIEZO1 in vessel‑lining cells is not just a bystander but a necessary driver of disease.
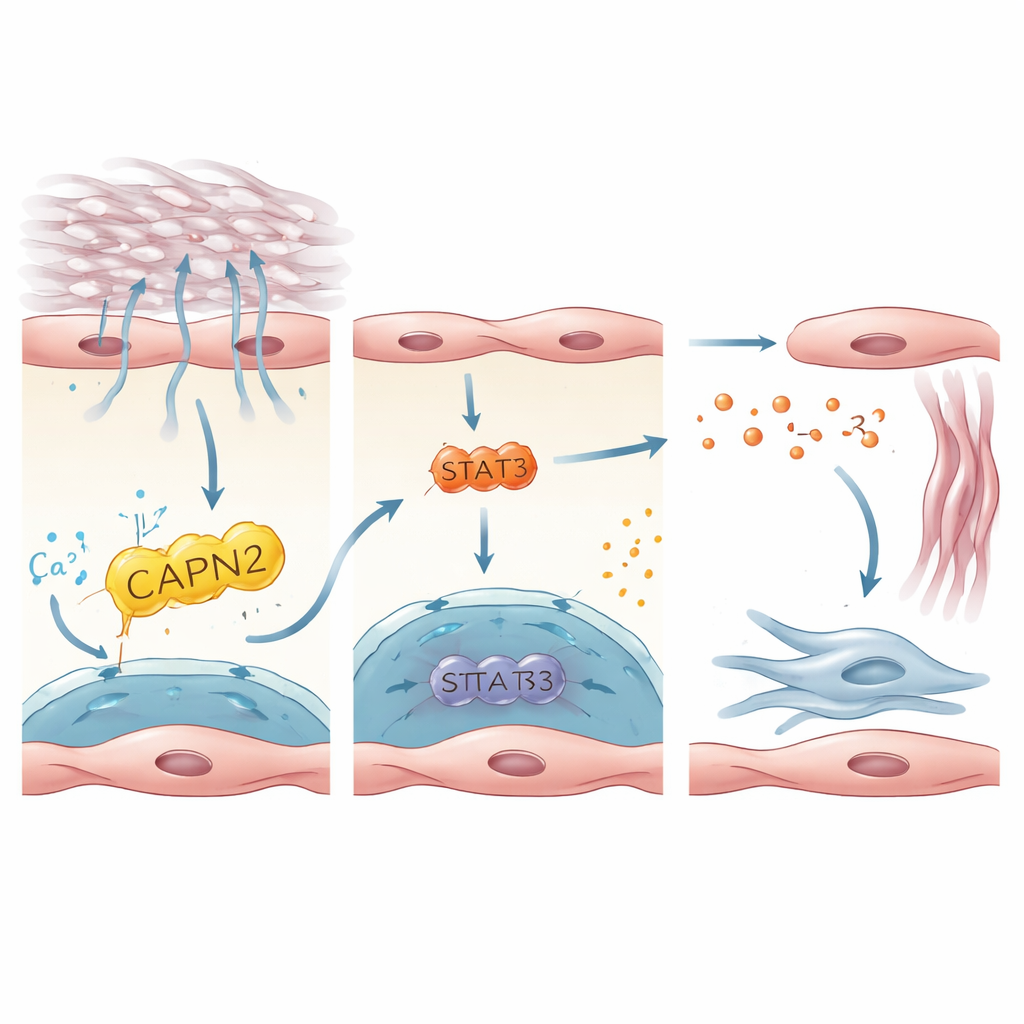
A Signaling Chain that Wakes Up Scar‑Forming Cells
The study then traced how PIEZO1 activation inside endothelial cells is translated into signals that awaken fibroblasts, the cells that lay down scar tissue. By integrating human and mouse datasets, the authors zeroed in on interleukin‑33 (IL‑33), a small protein released by stressed or damaged cells, as a key messenger. IL‑33 was strongly expressed in PIEZO1‑positive endothelial cells and elevated in lungs from patients and mice with fibrosis. In cultured human endothelial cells grown on stiff substrates or stretched to mimic breathing against a rigid lung, PIEZO1 activation boosted IL‑33 production. This depended on a downstream enzyme, CAPN2, and a transcription factor, STAT3, which together tuned IL‑33 gene activity. In mice, deleting IL‑33 specifically in endothelial cells protected against fibrosis, while forcing endothelial cells to overproduce IL‑33 erased the protective effect of losing PIEZO1. These results outline a linear axis: mechanical stress → PIEZO1 → CAPN2/STAT3 → IL‑33 → fibroblast activation and scarring.
What This Means for Future Treatments
For non‑specialists, the message is that lung fibrosis is not driven by rogue immune cells alone; it is also a disease of faulty “touch” in the blood vessels. Endothelial cells feel that their surroundings have grown too stiff, flip on the PIEZO1 switch, and in response release IL‑33, a danger signal that urges nearby fibroblasts to keep laying down scar. By dissecting this chain from mechanical force to gene expression, the work highlights several promising targets—PIEZO1 itself, the CAPN2‑STAT3 relay, and IL‑33—for therapies aimed at interrupting the self‑reinforcing cycle of stiffness and scarring. While more studies are needed to safely modulate these pathways in people, this mechanosensitive endothelial axis offers a new, physically grounded angle for tackling a disease that has long resisted effective treatment.
Citation: Zhang, L., Gui, X., Hou, R. et al. Single-cell multiomics uncovers an endothelial mechanosensitive PIEZO1-IL-33 axis driving pulmonary fibrosis. Nat Commun 17, 2655 (2026). https://doi.org/10.1038/s41467-026-70193-w
Keywords: pulmonary fibrosis, endothelial cells, mechanotransduction, PIEZO1, IL-33