Clear Sky Science · en
Multi-modal dissection of cell-type specific TDP-43 pathology in the motor cortex
Why this research matters for people
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are devastating brain diseases that rob people of movement, speech, and personality. Most patients with ALS and many with FTD share a common microscopic hallmark: clumps of a protein called TDP-43 building up where they do not belong. This study asks two practical questions with big implications for future treatments: exactly which brain cells are hit hardest by TDP-43 problems, and what goes wrong inside those cells at the level of DNA regulation and gene activity?
Following the damage into the brain's movement center
The researchers focused on the primary motor cortex, the strip of brain tissue that controls voluntary movement. Using post-mortem donated brain samples from people with ALS, ALS-FTD, and neurologically healthy controls, they isolated individual cell nuclei and read out both which genes were active and how tightly the local DNA was packed. This “multi-omic” approach, applied to more than 180,000 nuclei, allowed them to sort cells into precise types: several classes of excitatory and inhibitory neurons, as well as support cells such as astrocytes, oligodendrocytes, and microglia. They then combined this with spatial gene maps from another human brain dataset to place these cell types back into the familiar layered structure of the cortex.
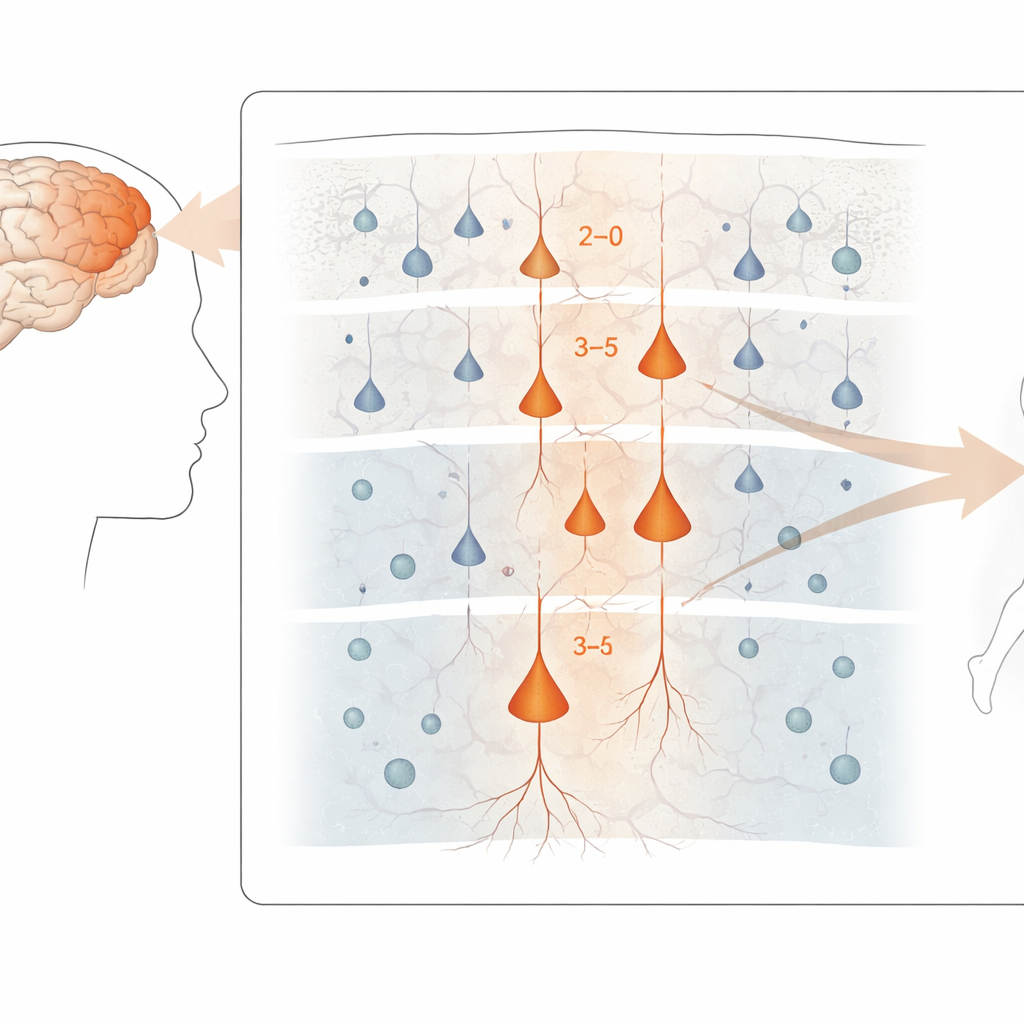
Pinpointing the most vulnerable neurons
Across the motor cortex, the strongest disease-related gene changes appeared in excitatory neurons, the cells that drive activity forward along brain circuits. In particular, upper and middle layer neurons that connect within the cortex, as well as certain deep-layer cells that send signals out of the cortex—including the large “Betz” cells that control spinal motor neurons—showed the most pronounced alterations. In contrast, inhibitory interneurons and many glial cells were less affected at the gene-expression level, even though some of them did show subtler shifts. Despite this molecular turmoil, the overall mix of major cell types in the tissue was surprisingly similar between patients and controls, suggesting that the damage is more about how cells function than simply how many are lost.
How TDP-43 reshapes gene activity from within
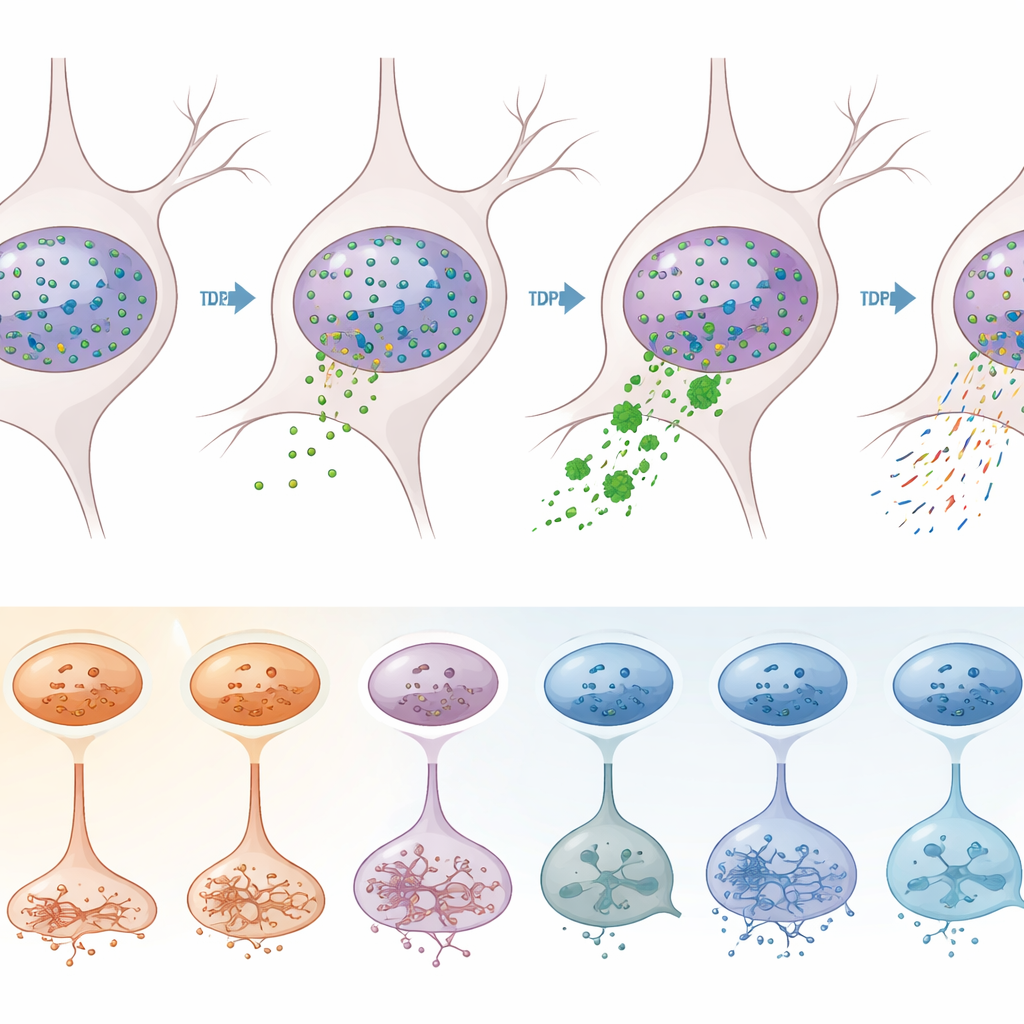
To separate effects driven directly by TDP-43 from other disease processes, the team used a clever sorting strategy. They labeled nuclei with antibodies against TDP-43 and a neuronal marker, then used flow cytometry to separate neurons whose nuclei had lost TDP-43 (a sign of pathology) from those that retained it. Sequencing more than 12,000 of these nuclei revealed that TDP-43 loss occurs overwhelmingly in excitatory neurons, especially specific subtypes in layers 2–3, 3–5, 5, and 6. In those vulnerable neurons, hundreds of genes were misregulated, including many already linked to ALS. Classic molecular signatures of TDP-43 malfunction—such as the appearance of “cryptic” extra pieces in STMN2 and KALRN gene transcripts, and shifts in where RNA molecules are cut and tailed at their ends—were clearly enriched in the TDP-43–deficient nuclei.
Epigenetic remodeling: not all change is from TDP-43
Because they measured both gene activity and chromatin openness in the same nuclei, the authors could ask which changes were associated with shifts in DNA packaging. They found tens of thousands of spots in the genome where local chromatin accessibility tracked with gene expression. Many of the genes altered in ALS and ALS-FTD lay in such regions, indicating that part of the disease signature reflects broader epigenetic remodeling rather than direct fallout from TDP-43 loss. Interestingly, these chromatin-linked changes often converged on signaling pathways involved in cell communication and axon guidance, and they were especially strong in certain excitatory neurons and oligodendrocytes. When the team compared gene changes tied to TDP-43 pathology with those tied to chromatin shifts, they saw that these were partly overlapping but largely distinct layers of disruption.
What this means for future therapies
For a lay reader, the key message is that ALS and ALS-FTD do not damage the motor cortex uniformly. Instead, they strike particular excitatory neuron types and, to a lesser degree, certain support cells, altering their gene programs in ways that depend both on the misbehavior of TDP-43 and on broader changes in how DNA is packaged and read. These findings suggest that effective treatments may need to be both cell-type specific and pathway specific—for example, restoring TDP-43 function or correcting its splicing errors in the most vulnerable neurons, while separately targeting epigenetic and signaling changes shared across multiple cell types. By mapping this complex landscape in high detail, the study provides a blueprint for designing more precise interventions aimed at slowing or preventing the loss of movement control in ALS and ALS-FTD.
Citation: Ruf, W.P., Kühlwein, J.K., Meier, L. et al. Multi-modal dissection of cell-type specific TDP-43 pathology in the motor cortex. Nat Commun 17, 2406 (2026). https://doi.org/10.1038/s41467-026-69944-6
Keywords: ALS, frontotemporal dementia, TDP-43, motor cortex neurons, single-nucleus multiomics