Clear Sky Science · en
Context-dependent translation inhibition as a cancer therapeutic modality
Turning Protein Factories Against Cancer
Cancer cells survive by constantly making large amounts of short‑lived "driver" proteins that keep them growing and dividing. This study explores a new way to fight cancer by subtly jamming the cell’s protein‑making machines—ribosomes—only when they try to build particular protein sequences, especially those found in hard‑to‑drug oncogenes like MYC. That selective interference could kill tumor cells while sparing much of normal protein production.
How Cells Build Proteins—and Where Things Can Go Wrong
Every cell depends on ribosomes, tiny molecular factories that read genetic messages (mRNAs) and string together amino acids into proteins. Most existing drugs that target ribosomes, such as antibiotics or older cancer drugs, act like blunt instruments: they broadly shut down protein synthesis, which can damage healthy cells and cause serious side effects. The authors reasoned that because each nascent protein chain has its own unique sequence and chemical character, it might be possible to design small molecules that only block the ribosome when a specific sequence is in the machine, leaving the rest of the cell’s protein production largely intact.
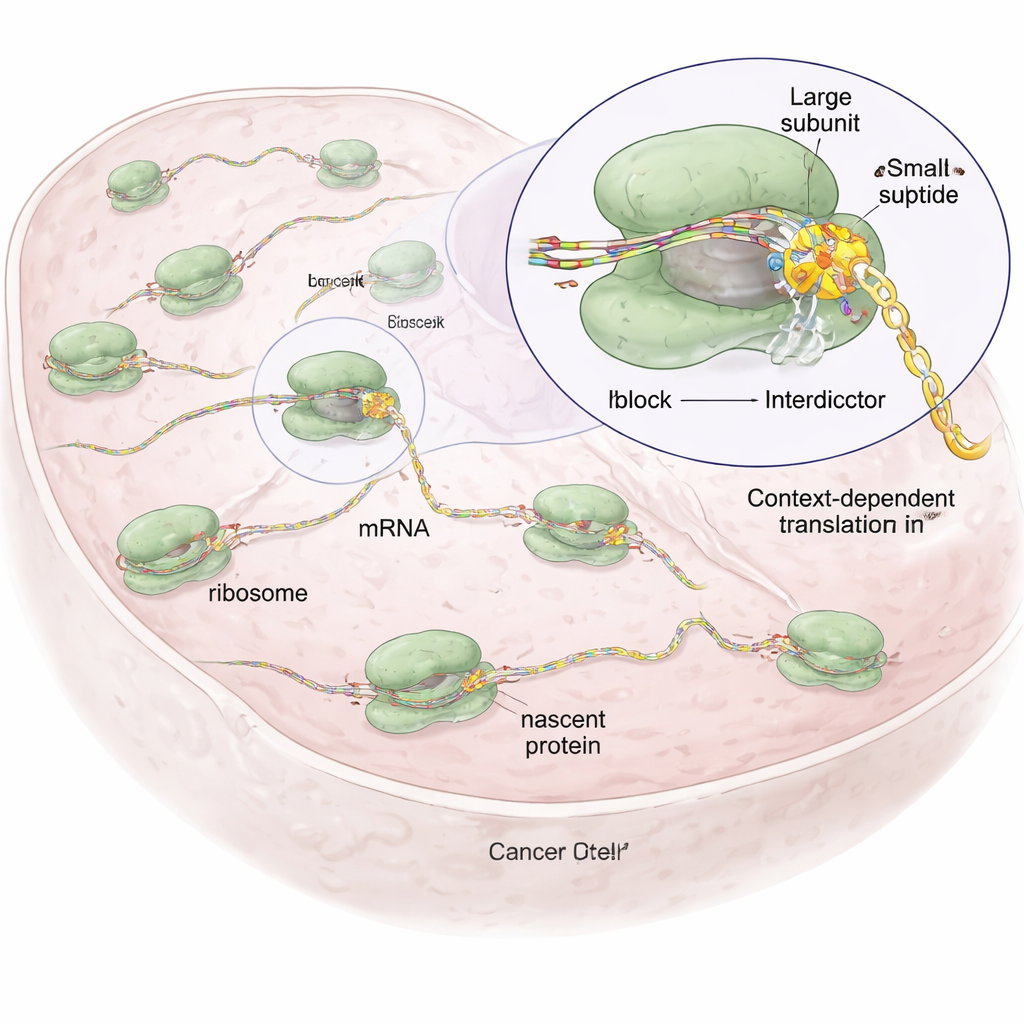
Designer Molecules That Stall Only Certain Protein Sequences
Building on a natural compound called anisomycin, which binds to the catalytic heart of the ribosome where peptide bonds are formed, the researchers created a family of synthetic molecules they call “interdictors.” All share a core scaffold that anchors them inside the ribosome, but they differ in side groups that point toward the growing protein chain. Those side groups are tuned to favor particular types of amino acids—for example, one interdictor (IDB‑001) is attracted to negatively charged residues, while another (IDB‑002) prefers small, oily (hydrophobic) residues. Using a technique called ribosome profiling, which globally maps where ribosomes pause on mRNAs inside cells, the team showed that each compound causes stalls at distinct short motifs in the nascent chain, often at the penultimate (−1) amino acid, demonstrating sequence‑dependent action rather than indiscriminate shutdown.
Watching the Drug and the Ribosome Meet Atom by Atom
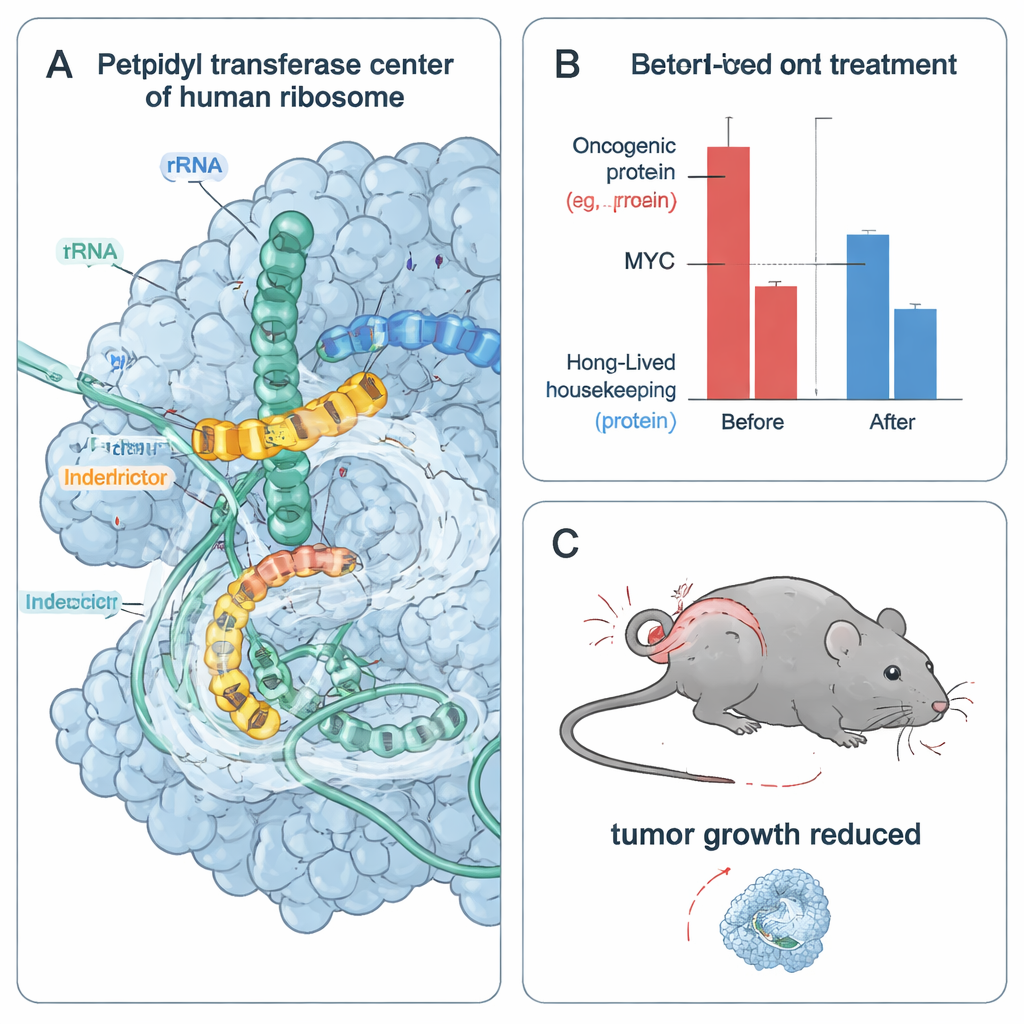
To see how this selectivity works in physical detail, the authors froze human ribosomes in the act of translating designed peptide sequences and imaged them by cryogenic electron microscopy at near‑atomic resolution. They observed the interdictor nestled in the ribosome’s active site making precise contacts with the last few amino acids of the nascent chain. In one structure, a hydrophobic side group of IDB‑002 is cradled by small non‑bulky residues in the peptide, explaining why larger side chains there are disfavored. In another, the acidic side chain of a MYC‑derived residue swings to form a salt bridge with a positively charged group on IDB‑001. The drug’s presence also nudges nearby ribosomal RNA bases into new positions that constrict the active site and partially block entry of the next tRNA, helping to freeze elongation at those favored sequences.
From Stalled Ribosomes to Stressed and Dying Tumor Cells
Because rapidly dividing cancer cells rely heavily on short‑lived oncogenic proteins such as MYC and CCND1, the team tested how interdictors affect tumor cell survival. In multiple MYC‑dependent cancer cell lines, IDB‑001 and IDB‑002 reduced cell viability at nanomolar to low‑micromolar concentrations. A further optimized analog, IDB‑003, was even more potent and suitable for oral dosing. In cells, these compounds rapidly depleted MYC and other fast‑turnover oncoproteins while leaving longer‑lived housekeeping proteins relatively stable over the same time window. The drugs also triggered cellular stress responses linked to collided ribosomes, but blocking those stress‑signaling pathways did not eliminate the loss of viability, suggesting that direct deprivation of essential oncogenic proteins is a major driver of tumor cell death.
Proof of Concept in a Difficult Breast Cancer Model
To test whether this approach works in animals, the authors treated mice bearing human triple‑negative breast cancer tumors with orally delivered IDB‑003. Over 28 days, treated tumors grew far more slowly than those in control animals, with up to 80% tumor growth inhibition at higher doses and no reported severe toxicity in this study. Gene‑expression analysis of the tumors showed strong downregulation of MYC target gene programs, consistent with reduced MYC activity in vivo. Together, these results indicate that selectively stalling ribosomes on specific protein sequences can weaken tumors that depend on unstable oncogenic drivers while potentially avoiding the broad toxicity of classical translation blockers.
Why This Matters for Future Cancer Treatments
This work introduces a new kind of small‑molecule drug: one that acts not by binding a finished protein, but by intercepting it as it is being made and only when a short “address tag” sequence is present. Because many cancer‑promoting proteins are floppy, short‑lived, or lack obvious pockets for conventional drugs, targeting their synthesis directly at the ribosome could open a path to treating tumors driven by currently "undruggable" genes like MYC. The study also suggests that by tuning the chemistry of interdictors, future medicines might be designed to focus on different sequence motifs and disease targets, extending this strategy beyond oncology to other conditions where dialing down specific proteins could restore health.
Citation: Diamond, P.D., Sauer, P.V., Holm, M. et al. Context-dependent translation inhibition as a cancer therapeutic modality. Nat Commun 17, 1963 (2026). https://doi.org/10.1038/s41467-026-69891-2
Keywords: ribosome-targeted cancer therapy, translation inhibition, MYC-driven tumors, context-dependent small molecules, triple-negative breast cancer