Clear Sky Science · en
Radiolabeling of oligopeptides by selective hydrogen isotope exchange with deuterium and tritium in aqueous buffers
Tracing Medicines at the Atomic Level
Modern medicines increasingly include complex biological molecules such as peptides and small proteins. To understand where these drugs go in the body and how long they last, scientists often swap a few ordinary atoms for rare or radioactive ones that can be tracked. This paper presents a way to “tag” peptide drugs with such traceable atoms directly in water-based solutions, much closer to real biological conditions than most earlier methods.
Why Tiny Atomic Swaps Matter
Replacing normal hydrogen with heavier forms like deuterium or tritium turns everyday molecules into powerful scientific tracers. These labeled versions behave almost the same as the original drug but can be followed by sensitive instruments that detect mass or radiation. For small drug molecules, chemists have built a large toolbox to make such labeled compounds. In contrast, methods for labeling larger, fragile biologics—such as peptides and proteins—are scarce, often complicated, and poorly suited to water-based environments similar to blood or cell fluids. The authors set out to solve this gap: a simple, selective way to insert deuterium or tritium into peptide building blocks directly in aqueous buffers.
A One-Pot Labeling Strategy in Water
The team focused on a type of reaction called hydrogen isotope exchange, in which a hydrogen atom on a molecule is swapped for its heavier cousin from a gas like deuterium (D2) or tritium (T2). They built an in situ catalyst based on iridium metal and a specially chosen phosphine helper molecule. When mixed in a mildly basic buffer and heated, this system activates specific carbon–hydrogen bonds on amino acids and small peptides and replaces those hydrogens with deuterium or tritium from the gas. Crucially, this is done in a single step, in water-rich media, and with very low amounts of metal—conditions that are friendlier to delicate peptide drugs and to practical laboratory workflows. 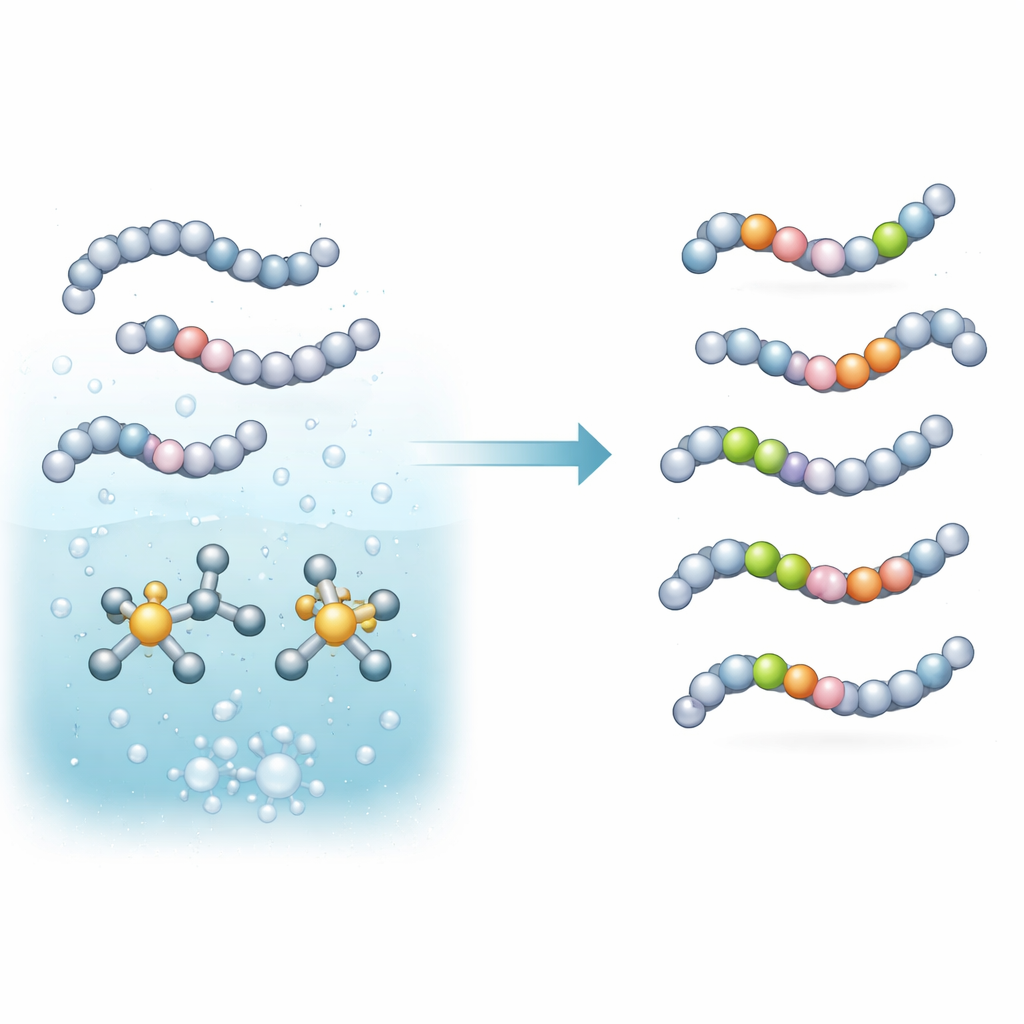
Picking Just the Right Spots on Peptides
Not every hydrogen atom in a peptide is equally useful as a label. Some are easily lost during metabolism, which would erase the radioactive tag. The authors carefully explored where their catalyst prefers to act. They discovered that unprotected amino acids such as lysine and arginine are particularly well suited. In lysine, the method selectively labels a carbon in the side chain (the so-called gamma position), a site considered “non-activated” and more likely to remain stable in the body. Arginine shows similar behavior at nearby positions on its side chain. By testing a series of related molecules, including short chains with two amino groups, the team found that having two nitrogen sites positioned just right helps the metal catalyst clamp onto the molecule and reach the targeted carbon–hydrogen bond.
Peering Under the Hood of the Catalyst
To understand why this selectivity arises, the researchers combined experiments with detailed computer simulations using density functional theory. These calculations map out the energy landscape as the iridium complex is formed from a dimeric starting material, binds to water, then to the amino acid, and finally inserts into a specific carbon–hydrogen bond. The models show that breaking the original iridium dimer is energetically feasible in water for one type of precursor but not for a closely related one, explaining why only certain starting complexes are effective. They also reveal that the substrate itself helps stabilize the active metal center and prevent it from clumping into inactive particles. The most favorable pathway involves the amino acid binding through two nitrogen atoms, forming a “pincer-like” grip that positions a single carbon–hydrogen bond for exchange with deuterium or tritium.
From Simple Building Blocks to Real Peptide Drugs
With the mechanism in hand, the team extended the method from single amino acids to short peptides containing up to seven residues and then to more complex therapeutic-like sequences with as many as 13 amino acids. In all cases, the labeling occurred at the side chains of lysine or arginine at the peptide’s end, and the peptides remained largely intact under the reaction conditions. For tritium, they optimized the reaction at low gas pressures to safely achieve high specific activities, meaning that a large fraction of molecules carry at least one tritium atom. These tritium-labeled peptides were produced in one pot and are ready for use as tracers in in vitro and potentially in vivo studies. 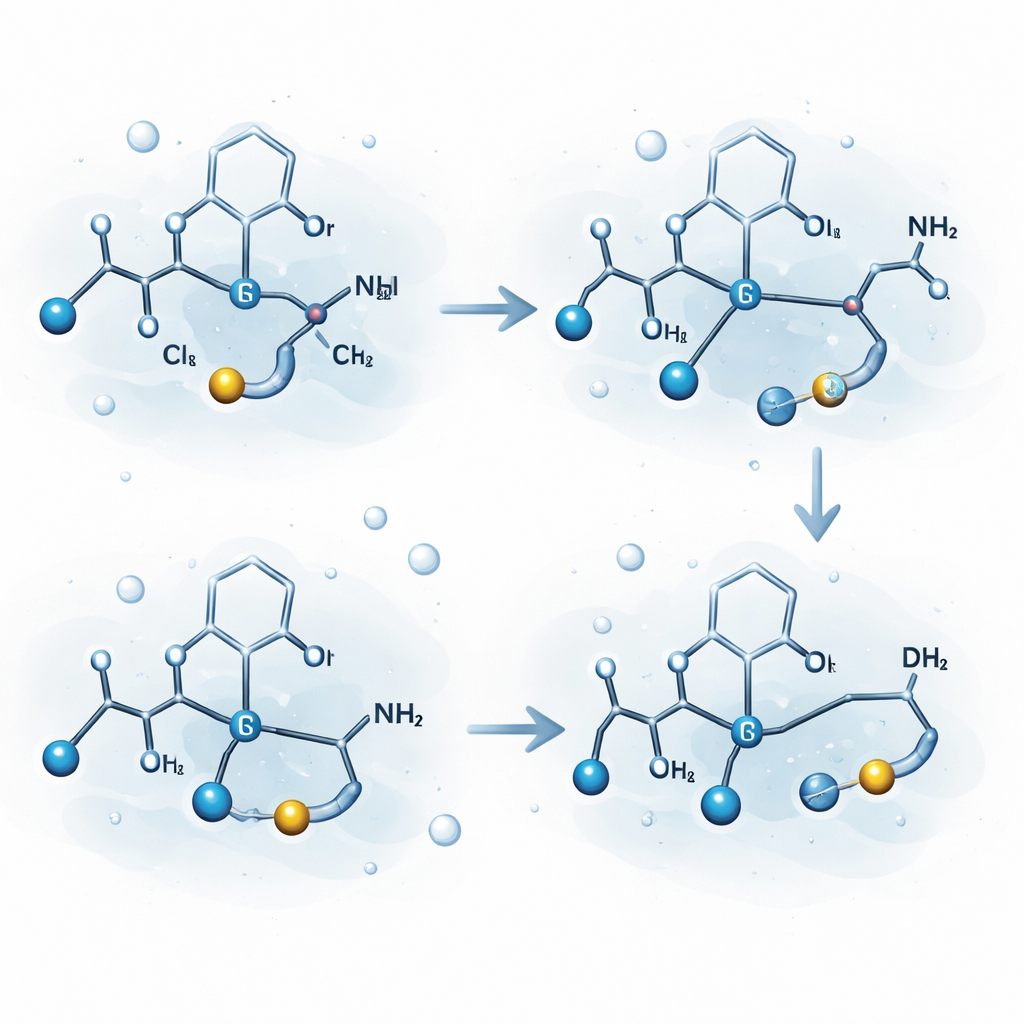
What This Means for Future Medicines
This work demonstrates that it is possible to selectively attach deuterium or tritium to realistic peptide drugs in a simple, water-based step while targeting metabolically robust positions on key amino acids. For drug developers, that means easier access to precisely labeled tracer versions of peptide therapies, which are vital for measuring absorption, distribution, and metabolism. Beyond tracer production, the mechanistic insights into how the iridium catalyst interacts with amino acids may inspire new ways to fine-tune where and how complex biomolecules are modified, opening doors to more precise chemical control over future biologic medicines.
Citation: Martinelli, E., Weck, R., Güssregen, S. et al. Radiolabeling of oligopeptides by selective hydrogen isotope exchange with deuterium and tritium in aqueous buffers. Nat Commun 17, 2317 (2026). https://doi.org/10.1038/s41467-026-69850-x
Keywords: radiolabeled peptides, hydrogen isotope exchange, deuterium and tritium labeling, peptide therapeutics, iridium catalysis