Clear Sky Science · en
Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes
Watching Proteins in Motion
Many of the molecules that keep us alive behave less like rigid Lego bricks and more like tiny machines that constantly change shape. These motions power processes such as making energy, repairing DNA, and letting viruses enter cells. Experiments like cryo–electron microscopy can now freeze some of these shapes, but not the fleeting steps in between. This article introduces eBDIMS2, a new computer method that can "fill in the missing frames" of protein motion even for huge molecular machines that were previously too large and complex to simulate on an ordinary computer.
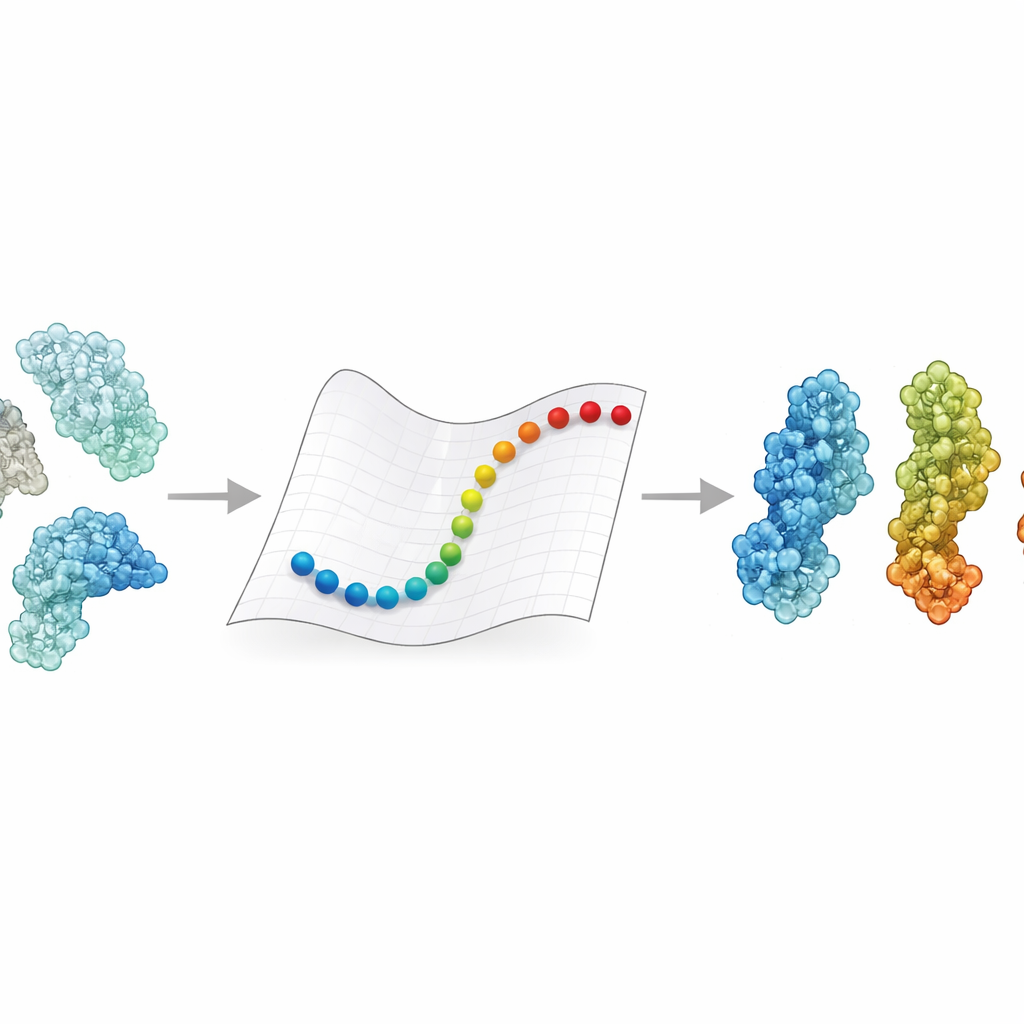
Why Protein Shape Changes Matter
Proteins rarely stay locked in a single pose. They open and close, twist and bend in response to signals like voltage changes, pH, or the binding of a partner molecule. These shifts can mean the difference between an enzyme being on or off, or a receptor catching a virus or letting it slip away. Experiments give us detailed snapshots of a few key shapes, and molecular dynamics simulations can in principle connect them by following every atom over time. But tracking such motion for the huge complexes now seen by cryo–electron microscopy, often weighing hundreds of thousands to millions of Daltons, usually demands supercomputers and weeks of processing. As a result, for many medically important giants, we still do not know how one state transforms into another.
A Faster Route Through Protein Landscapes
eBDIMS2 takes a shortcut by simplifying how proteins are represented and how their motion is calculated. Instead of following every atom, it treats each amino acid as a single point connected by springs in an elastic network. These springs capture how different parts of the protein tend to move together. The method then uses Brownian dynamics—mathematical rules that mimic jiggling in a liquid—to nudge the structure from one experimentally known state toward another. Crucially, eBDIMS2 only pays attention to interactions that really matter, using distance cutoffs and parallel computation to reduce the cost. This improves the program’s scaling from roughly quadratic to almost linear with protein size. In practice, it means that transitions for enormous assemblies approaching two million Daltons can be explored in hours on a desktop, instead of being essentially unreachable.
Checking the Paths Against Real Proteins
To see whether these fast paths make biological sense, the authors assembled ensembles of 47 large proteins and 15 additional complexes, totaling hundreds of structures mostly solved by cryo–electron microscopy. They used principal component analysis, a statistical tool that picks out the dominant ways each protein can move, to organize these structures into landscapes of conformations such as open, closed, active, or inactive. eBDIMS2 was then asked to connect pairs of end states across this landscape. The resulting paths were projected back onto the same low–dimensional maps, revealing whether they trace smooth routes that pass near experimentally observed intermediates. In more than 30% of the systems, the simulated routes ran close—within a few ångströms—to intermediate structures that had not been provided as input. For demanding cases like the DNA repair enzyme DNA-PKcs or the coronavirus spike protein, the coarse-grained paths also overlapped well with much more expensive atom-level simulations, including targeted molecular dynamics and advanced enhanced-sampling runs.
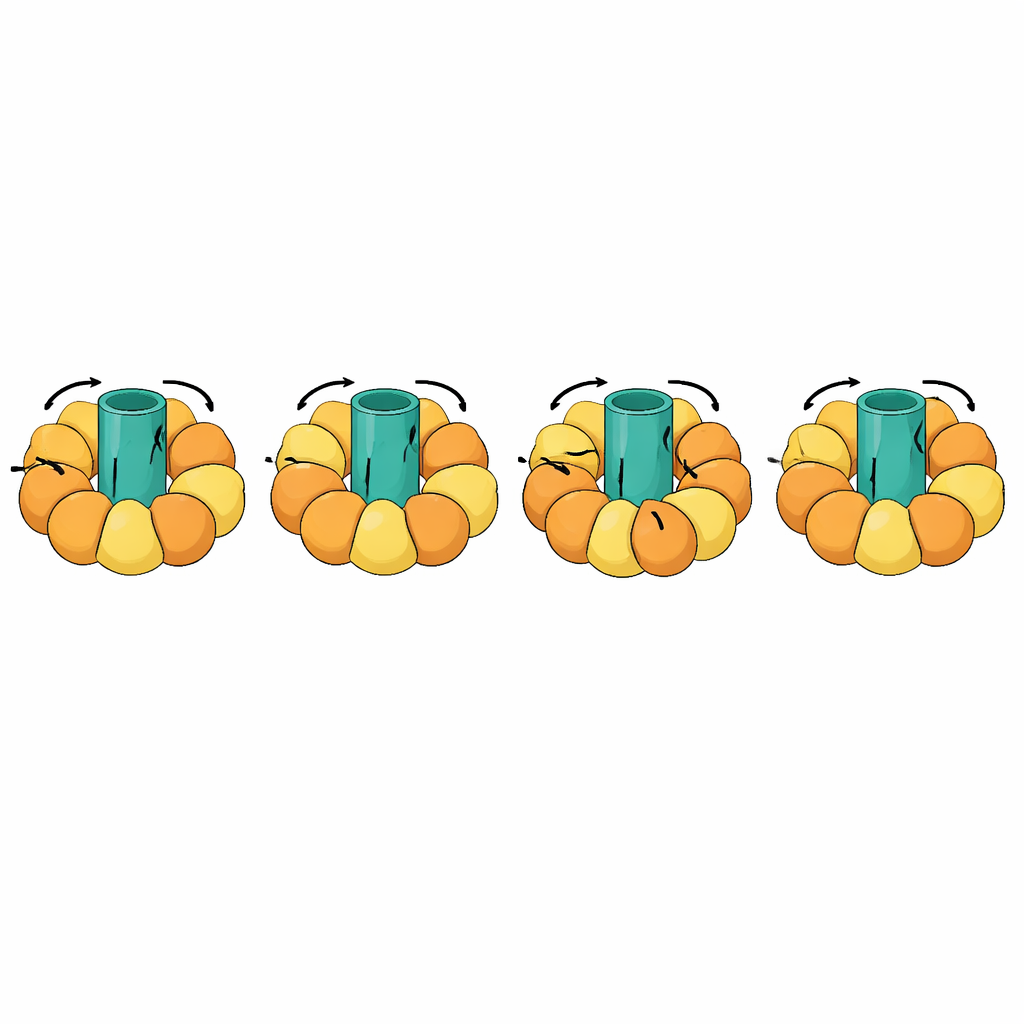
Following Giant Molecular Machines
One of the most striking tests involved rotary machines such as ATP synthases, which make the cell’s energy currency by coupling a spinning rotor in the membrane to opening and closing motions in surrounding subunits. These transitions are exceptionally complex: parts of the molecule must remain rigid and rotate as a unit, while others flex in a choreographed cycle. eBDIMS2 introduces special handling for such quasi-rigid pieces and for incomplete experimental models with missing segments, both common in cryo–electron microscopy. With these features, it can simulate full rotational cycles of ATP synthase and other massive complexes like molecular chaperones, receptors, and viral assemblies. Throughout, the generated intermediate structures avoid the severe distortions produced by some competing methods and can be cleaned up into atomistic models suitable for drug-design calculations or longer, more detailed simulations.
What This Means for Biology and Medicine
The study shows that eBDIMS2 can reliably sketch out the main routes between known protein shapes for systems that were out of reach for traditional simulations. It does not replace detailed atom-level movies or provide precise energies and timing, but it offers a fast, physically grounded way to map how large molecular machines might move, using only a pair of experimental structures as input. As structural databases fill with multiple states of big protein assemblies linked to cancer, infection, and other diseases, this approach gives researchers an accessible tool to connect the dots, suggest plausible intermediate states, and guide where to look next with higher-resolution methods or targeted drug design.
Citation: Scaramozzino, D., Lee, B.H. & Orellana, L. Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes. Nat Commun 17, 2202 (2026). https://doi.org/10.1038/s41467-026-69809-y
Keywords: protein dynamics, molecular simulations, cryo-EM, conformational pathways, coarse-grained modeling