Clear Sky Science · en
Activation of IRF3 in cardiomyocytes impairs mitochondrial oxidative function through PGC-1α inhibition and drives heart failure
Why stressed hearts and tired cells matter
Heart failure is often described as the heart “wearing out,” but under the hood it is also a story about chronic inflammation and exhausted power plants inside heart muscle cells. This study asks a deceptively simple question with big implications: is there a single molecular switch inside heart cells that ties together harmful inflammation and failing energy production—and if so, can flipping that switch change the course of heart failure? By following that thread, the authors uncover a key player and show that gently boosting the heart’s own energy program can partly rescue failing hearts in mice.
A molecular switch in sick human hearts
The researchers focused on a protein called IRF3, best known for helping cells respond to viral infections. They examined tissue from people with ischemic cardiomyopathy, a common form of heart failure caused by reduced blood flow after heart attacks. In these failing hearts, IRF3 was not just present—it was chemically switched on at specific sites, a sign that it was actively driving gene programs. At the same time, the machinery that lets mitochondria turn fuel into energy through oxidative phosphorylation was noticeably weakened. A similar pattern appeared in mouse models of heart attack: when a coronary artery was tied off, IRF3 in heart muscle cells became strongly activated, and genes controlled by IRF3 lit up. Even fragments of mitochondrial DNA—released from damaged mitochondria and acting as internal “danger” signals—were enough to switch on IRF3 in isolated heart cells.
Turning IRF3 off protects the heart
To test whether IRF3 activity in heart muscle cells actually worsens disease, the team engineered mice in which IRF3 could be removed only from cardiomyocytes, leaving other immune and support cells untouched. After inducing a heart attack, these mice had better pumping function and less scarring than normal mice, despite having the same initial injury. In heart cells grown in dishes, silencing IRF3 toned down inflammatory genes without disrupting other related proteins. Together, these results argue that IRF3 inside the heart cell itself is not just a bystander: it amplifies inflammation and structural damage after ischemia and helps drive the transition to heart failure.
When IRF3 is stuck “on,” the fuel system collapses
The authors then flipped the experiment: they created mice in which IRF3 in cardiomyocytes could be forced into a permanently active state using a clever genetic “phosphomimetic” trick. Even without an external trigger, these mice rapidly developed severe heart dysfunction, high levels of inflammatory messengers in the blood, and signs of cellular injury. A deep dive into their heart tissue showed that, when IRF3 is chronically active, it suppresses a master energy coordinator called PGC-1α. This molecule normally promotes healthy mitochondria, efficient burning of fats, and balanced cellular energy. With PGC-1α pushed down, multiple mitochondrial proteins fell, the electron transport chain faltered, and the heart’s fuel choices shifted: carnitine and related compounds for fat burning dropped, ketone use was impaired, and glucose handling became distorted. Even the ratio of NAD⁺ to NADH—a key indicator of cellular redox balance—tilted in the wrong direction.
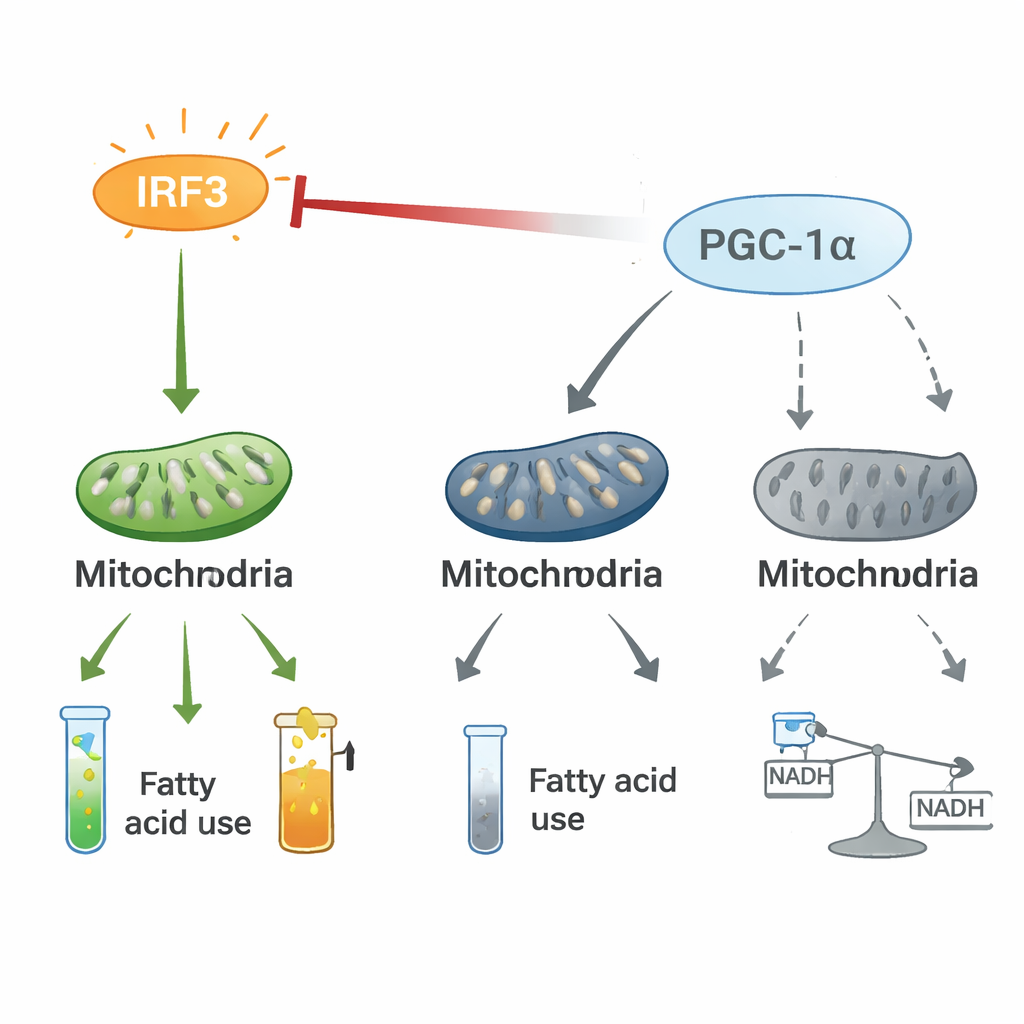
A tug-of-war between inflammation and energy control
Mechanistic experiments revealed that IRF3 and PGC-1α form a two-way regulatory axis. In heart cells, activated IRF3 physically associates with PGC-1α and blunts its ability to turn on fat-burning genes. Knocking down IRF3 raises PGC-1α levels and activity, while boosting PGC-1α dampens IRF3-driven inflammatory genes and restores mitochondrial markers, even under stress conditions such as low oxygen or bacterial toxins. Stable isotope tracing showed that IRF3 activation reroutes carbon from normal energy production through the citric acid cycle into an alternative pathway, the pentose phosphate pathway, and disrupts the smooth flow of metabolites. This tug-of-war between a pro-inflammatory switch (IRF3) and an energy co-pilot (PGC-1α) appears to reshape the heart’s metabolism in ways that favor inflammation and energy loss.
Gently recharging the heart’s batteries
Finally, the team asked whether nudging PGC-1α back up could counteract IRF3’s damage. They used a heart-targeted gene therapy vector to raise PGC-1α moderately—but not excessively—in the same mice with hyperactive IRF3. This modest boost improved pumping function, increased mitochondrial proteins, enhanced genes for fat burning and NAD metabolism, and reduced inflammatory and fibrotic gene activity. In cell experiments, co-expressing PGC-1α with active IRF3 restored a healthier NAD⁺/NADH balance and shifted fuel use back toward fats. For a lay reader, this means that carefully recharging the heart’s “battery management system” can partially offset the harmful effects of a chronic inflammatory switch stuck in the “on” position.
What this means for future heart failure care
This work positions IRF3 as a central link between inflammation and energy failure inside heart muscle cells. Instead of treating inflammation and metabolism as separate problems in heart failure, the study suggests they are intertwined through an IRF3–PGC-1α axis. While these findings are in mice and cells, they raise the possibility that future therapies could either dial down IRF3 activity or bolster PGC-1α and mitochondrial function to slow or prevent heart failure after a heart attack. In simple terms, calming an overactive cellular alarm system and supporting the heart’s energy factories may prove to be a powerful combined strategy to keep weakened hearts beating strongly for longer.
Citation: Kumari, M., Evangelakos, I., Deshpande, A. et al. Activation of IRF3 in cardiomyocytes impairs mitochondrial oxidative function through PGC-1α inhibition and drives heart failure. Nat Commun 17, 2051 (2026). https://doi.org/10.1038/s41467-026-69792-4
Keywords: heart failure, inflammation, mitochondria, cardiomyocytes, PGC-1α