Clear Sky Science · en
GCN5 drives MASLD progression through LXRα/SREBP1c signaling pathway–mediated de novo lipogenesis
Why this liver story matters
Metabolic dysfunction–associated steatotic liver disease (MASLD), formerly called non‑alcoholic fatty liver disease, now affects about one in four people worldwide. It often develops silently, yet can progress to scarring, liver cancer, and serious metabolic problems. This study uncovers a molecular “volume knob” that helps drive harmful fat buildup in the liver and shows how turning it down could both protect the liver and potentially make existing heart‑drug strategies safer.
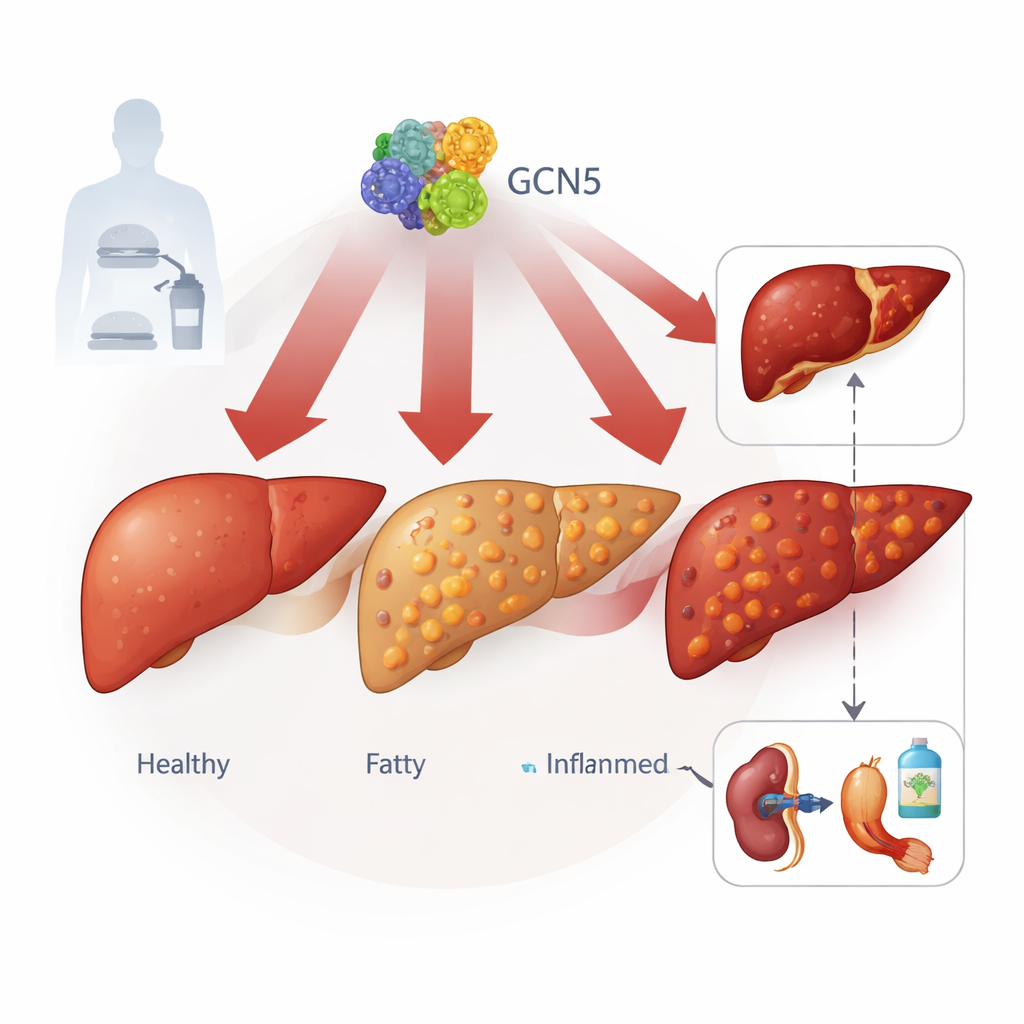
A hidden switch inside liver cells
The authors focus on a protein called GCN5, best known as a regulator of how tightly DNA is packaged. By studying liver tissue from more than 100 people, ranging from healthy to severe MASLD, and from several mouse models of diet‑induced fatty liver, they found that GCN5 levels in liver cells climb steadily as disease worsens. A closely related protein, PCAF, did not show this pattern, suggesting GCN5 plays a special role. High GCN5 tracked with more liver fat, higher blood lipids, and stronger signs of liver injury, linking this molecular switch to real‑world disease severity.
Turning GCN5 up or down in animals
To test cause and effect, the team genetically boosted or removed GCN5 specifically in mouse liver cells. When GCN5 was cranked up, mice on a high‑fat diet developed heavier, fattier livers, higher blood fats, and more liver cell damage, even though they did not eat more or gain extra body weight. Liver cells grown in dishes behaved similarly: extra GCN5 led to larger and more numerous fat droplets. By contrast, mice engineered to lack GCN5 only in their liver cells were strongly protected. Across several diet models that mimic human MASLD and its more severe inflammatory form, these animals accumulated less liver fat, showed lower blood lipids and liver enzymes, and developed less inflammation and scarring.
How GCN5 pushes the liver to make fat
Diving into metabolism, the researchers measured many fatty acids and their building blocks in the liver. Loss of GCN5 mainly reduced fats that the liver makes from scratch, a process known as de novo lipogenesis, while leaving diet‑derived polyunsaturated fats largely unchanged. Gene‑expression and isotope‑tracing experiments showed that GCN5 sits upstream of a master fat‑making regulator called SREBP1c. When GCN5 was active, genes that build and modify fatty acids were switched on, and the liver’s internal fat‑manufacturing rate rose. Removing or blocking GCN5 dialed this program down, cutting the flow of carbon from sugar into newly made liver fat.
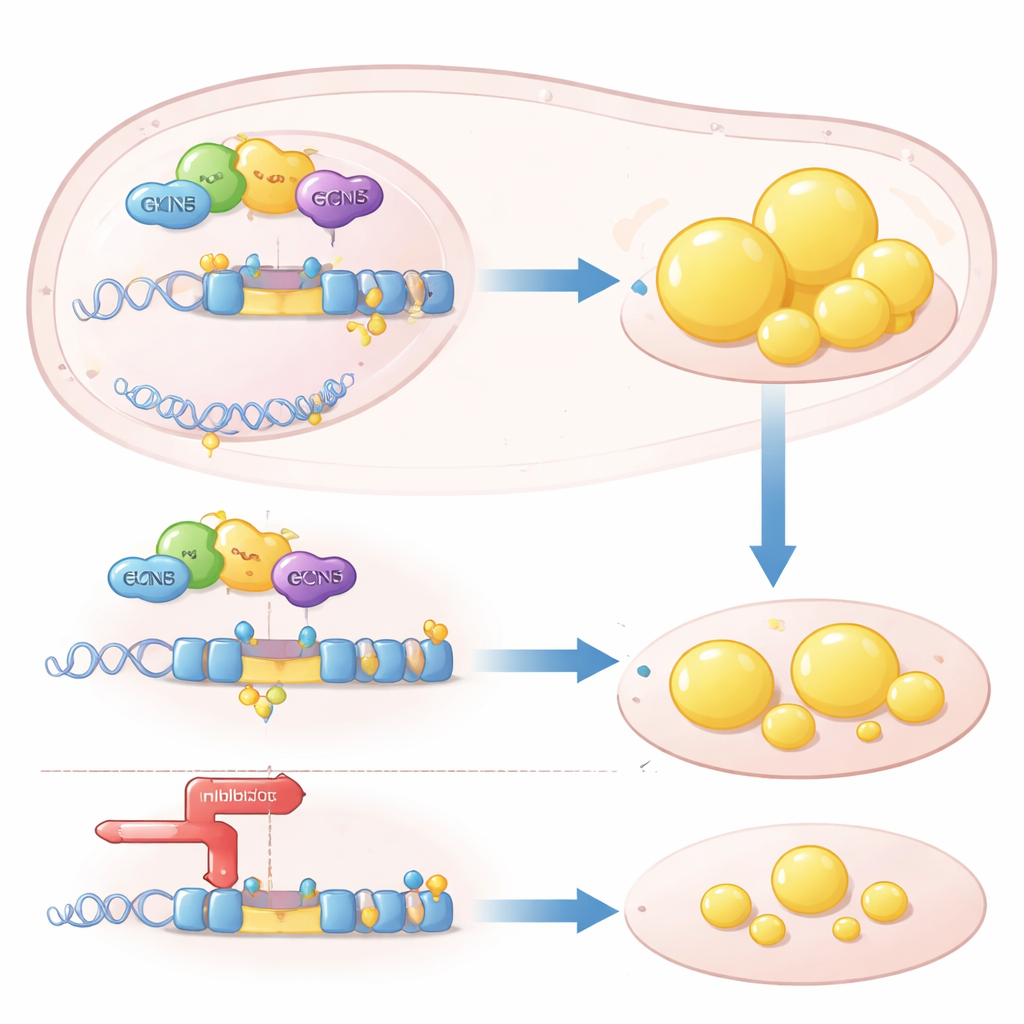
An epigenetic lever on a key fat gene
Mechanistically, GCN5 acts as an “epigenetic” lever: it chemically modifies histone proteins on DNA to make certain genes easier to read. The authors showed that GCN5 is recruited to the control region of the SREBP1c gene together with a nuclear receptor called LXRα, which senses cholesterol‑related molecules. Once there, GCN5 adds acetyl marks to histone H3, loosening the local chromatin and boosting SREBP1c transcription. This effect was highly selective: GCN5 enhanced LXRα’s ability to turn on SREBP1c but not another LXR target, ABCA1, which helps clear cholesterol from tissues. Without GCN5, LXRα could no longer efficiently engage the SREBP1c promoter, and the downstream fat‑synthesis program stalled.
A drug candidate and a promising combination
The team then tested CPTH2, a small‑molecule inhibitor of GCN5 that concentrates in the liver. In mouse models already placed on fatty diets, CPTH2 reduced liver size, fat content, and injury markers without obvious toxicity or changes in food intake. In cultured human and mouse liver cells, CPTH2 lowered fat droplets and triglycerides only when GCN5 was present, confirming that its action is specific. Importantly, in both cells and mice treated with LXR‑activating compounds (designed to improve cholesterol removal and fight atherosclerosis), CPTH2 selectively blocked the unwanted rise in SREBP1c‑driven fat production while preserving genes that promote reverse cholesterol transport. When combined with an LXR agonist in high‑fat‑fed mice, CPTH2 further lowered harmful blood lipids and liver cholesterol and prevented extra fat accumulation in the liver.
What this means for patients
The study positions GCN5 as a central driver of liver fat buildup in MASLD by linking dietary and hormonal signals to the SREBP1c fat‑production switch. Because GCN5 appears to be dispensable for the beneficial cholesterol‑clearing arm of LXR signaling, drugs that inhibit GCN5—such as CPTH2 or more advanced successors—could tame liver fat and inflammation while allowing heart‑protective therapies to work. For people at risk of both fatty liver disease and cardiovascular disease, targeting this epigenetic switch may one day offer a way to protect the liver without sacrificing the benefits of improving cholesterol handling.
Citation: Xiao, HT., Song, P., Jin, J. et al. GCN5 drives MASLD progression through LXRα/SREBP1c signaling pathway–mediated de novo lipogenesis. Nat Commun 17, 2821 (2026). https://doi.org/10.1038/s41467-026-69736-y
Keywords: fatty liver disease, epigenetics, lipid metabolism, liver metabolism, nuclear receptors