Clear Sky Science · en
Extending the range of graph neural networks with global encodings
Why long-distance links in molecules matter
From new medicines to better batteries, many of today’s breakthroughs rely on computer models that can predict how thousands of atoms push and pull on each other. A popular class of AI models, called graph neural networks, has become a workhorse for this task. Yet these models have a blind spot: they mostly pay attention to close neighbors, even though distant atoms can strongly influence one another through electric and quantum forces. This article introduces RANGE, a way to give these neural networks a kind of global view so they can "feel" and predict long-distance effects without becoming painfully slow or memory-hungry.
How current AI sees only the neighborhood
Graph neural networks treat a molecule or material as a web of nodes (atoms) linked by edges (their relationships). At each step, every node updates its state by talking only to its nearby neighbors within a fixed distance. Repeating this many times slowly spreads information, but this strategy has two big downsides. First, messages can get blurred as they pass through many intermediates, a problem known as oversmoothing. Second, narrow pathways in the graph can choke how much information gets through, causing oversquashing. Both issues become serious when trying to capture forces that act over many angstroms, such as electrostatics and dispersion in large molecules or crystals. Simply extending the interaction distance or stacking more layers makes the models expensive and still fails to fully solve these bottlenecks.
Adding virtual hubs for global communication
RANGE (Relaying Attention Nodes for Global Encoding) rewires this picture by adding a small set of virtual “master nodes” that do not correspond to any real atom. Instead, they act as global hubs. After an ordinary message-passing step between neighboring atoms, information from all atoms is gathered into these hubs using an attention mechanism: each master node learns which parts of the system to focus on. This aggregation creates coarse-grained summaries of the molecule’s state. In a second broadcast step, those summaries are sent back to every atom, again using attention so each atom can decide how much to listen to each master node while also keeping its local memory through self-loops. Because every atom connects directly to every master node, long-range communication can happen in a single step, turning the graph into a small-world network where distant regions can influence each other quickly and efficiently. 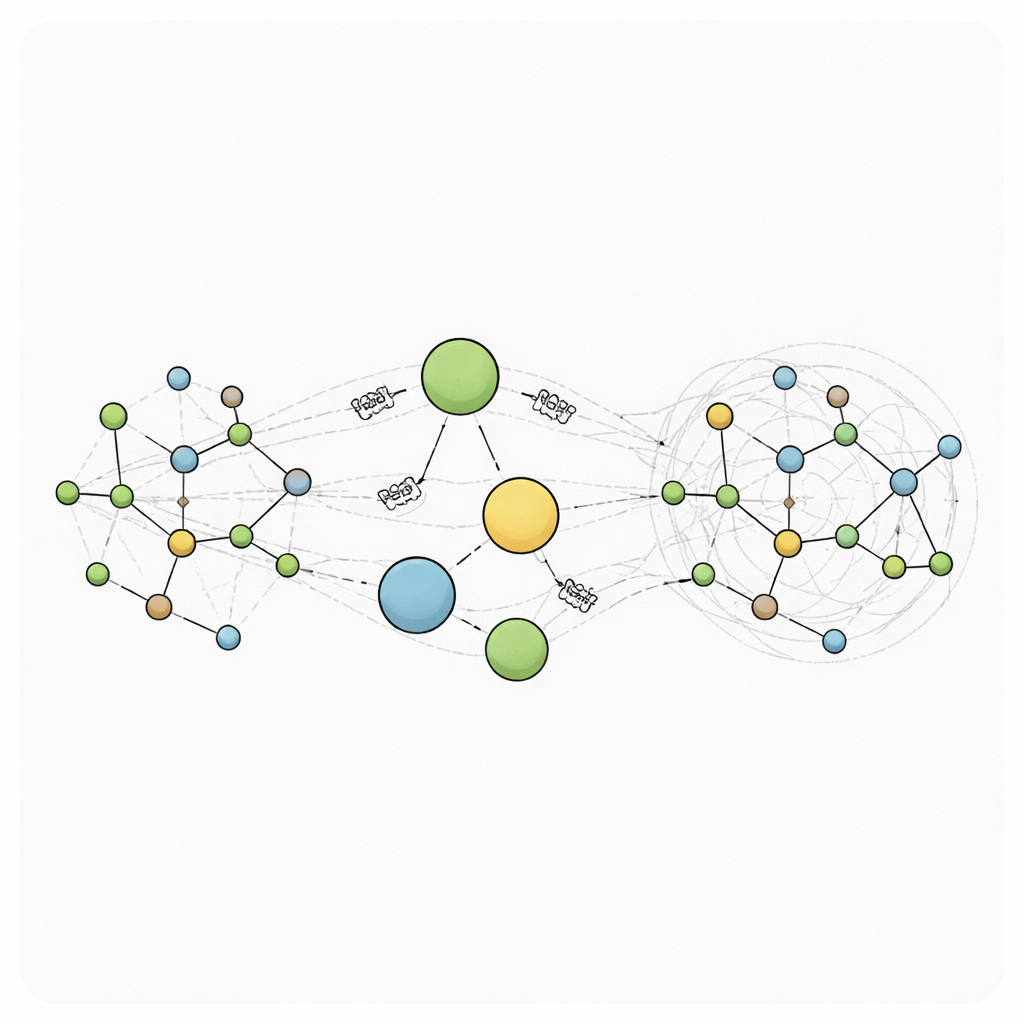
Seeing long-range forces that others miss
The researchers tested RANGE by attaching it to several state-of-the-art molecular force-field models and comparing them with their original, purely local versions. They used challenging systems where long-range effects are known to be crucial: a salt crystal with an extra sodium ion acting like a dopant, a gold dimer approaching a doped oxide surface, and pairs of organic molecules interacting at varying distances. Standard models largely failed to notice how distant charge rearrangements or hidden dopants changed the energy landscape; their predictions barely shifted when the long-range environment changed. In contrast, the RANGE-augmented models correctly captured the different energy curves and could extrapolate to larger separations than they had seen during training, with errors up to four times smaller for difficult charged dimers.
Accuracy without breaking the computer
Crucially, RANGE delivers this improved vision without the steep computational cost of other global approaches. Techniques that borrow from physics, like Ewald summation or Fourier-based corrections, require operations that grow roughly with the square of the number of atoms or depend on large grids, making them heavy for big systems and repeated simulations. RANGE, by design, scales linearly with system size: each master node connects to all atoms, but the number of master nodes grows modestly and is controlled by a regularization scheme that prevents them from becoming redundant. Benchmarks on larger datasets show that RANGE consistently reduces errors in predicted forces, even when the underlying models use short interaction cutoffs, and it does so with only a modest increase in runtime and memory. The team also ran molecular dynamics simulations for tens of nanoseconds on complex molecules, finding that RANGE-based force fields stayed stable and explored realistic shapes and states. 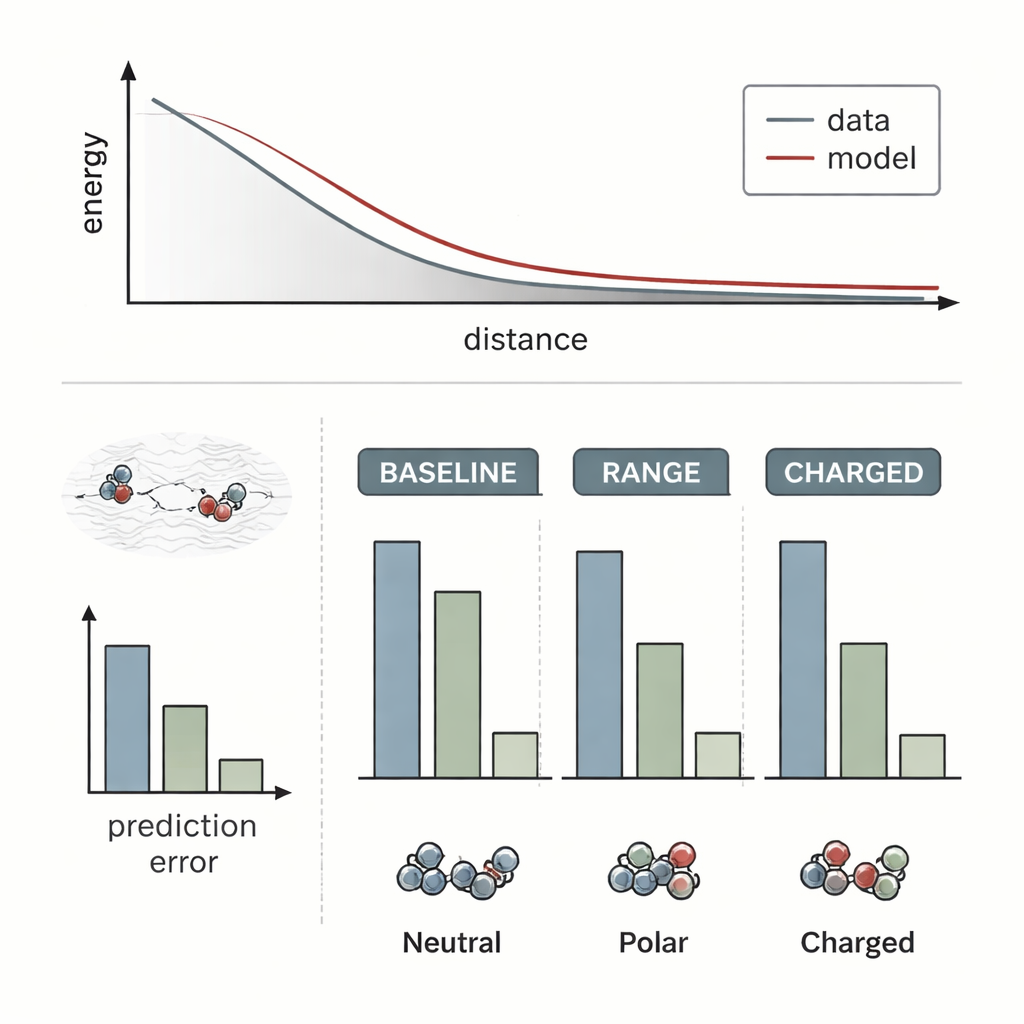
A clearer big-picture view of molecular worlds
For non-specialists, the key message is that RANGE gives existing graph-based AI models a new way to think globally while still operating locally. By introducing smart virtual hubs and attention-driven information flow, it overcomes the usual bottlenecks that prevent neural networks from capturing long-distance, many-body effects in molecules and materials. This means more reliable predictions for systems where far-apart regions subtly influence each other — from flexible drug molecules to extended nanostructures — without a prohibitive computational bill. As these methods are applied to ever larger and more complex environments, they promise AI tools that can more faithfully mirror the true, long-range fabric of physical interactions.
Citation: Caruso, A., Venturin, J., Giambagli, L. et al. Extending the range of graph neural networks with global encodings. Nat Commun 17, 1855 (2026). https://doi.org/10.1038/s41467-026-69715-3
Keywords: graph neural networks, long-range interactions, molecular simulations, machine-learned force fields, attention mechanisms