Clear Sky Science · en
Combination of PARP and KRASG12D inhibitors enhances therapeutic efficacy by exploiting vulnerabilities in PDAC
Why this study matters
Pancreatic cancer is one of the deadliest common cancers, largely because it is usually found late and shrugs off standard treatments. Many of these tumors are powered by a specific genetic glitch called KRASG12D, for which a new experimental drug shows promise but quickly runs into resistance. This study asks a practical question with real-world stakes: can we pair that KRAS-blocking drug with a second medicine to turn a short-lived response into a deeper, longer-lasting attack on the cancer?
A stubborn cancer with a common weak point
Most pancreatic ductal adenocarcinomas carry mutations in the KRAS gene, which acts like a stuck accelerator pedal for cell growth. Among these, the KRASG12D form is both the most frequent and the most closely tied to poor survival. The researchers first confirmed, using large cancer databases, that patients whose tumors carry this mutation tend to fare worse than those with other KRAS changes or none at all. They also noticed that KRASG12D tumors show high activity in genes that repair broken DNA, hinting that these cancers may depend on strong DNA-repair machinery to survive the constant damage that comes with rapid growth.
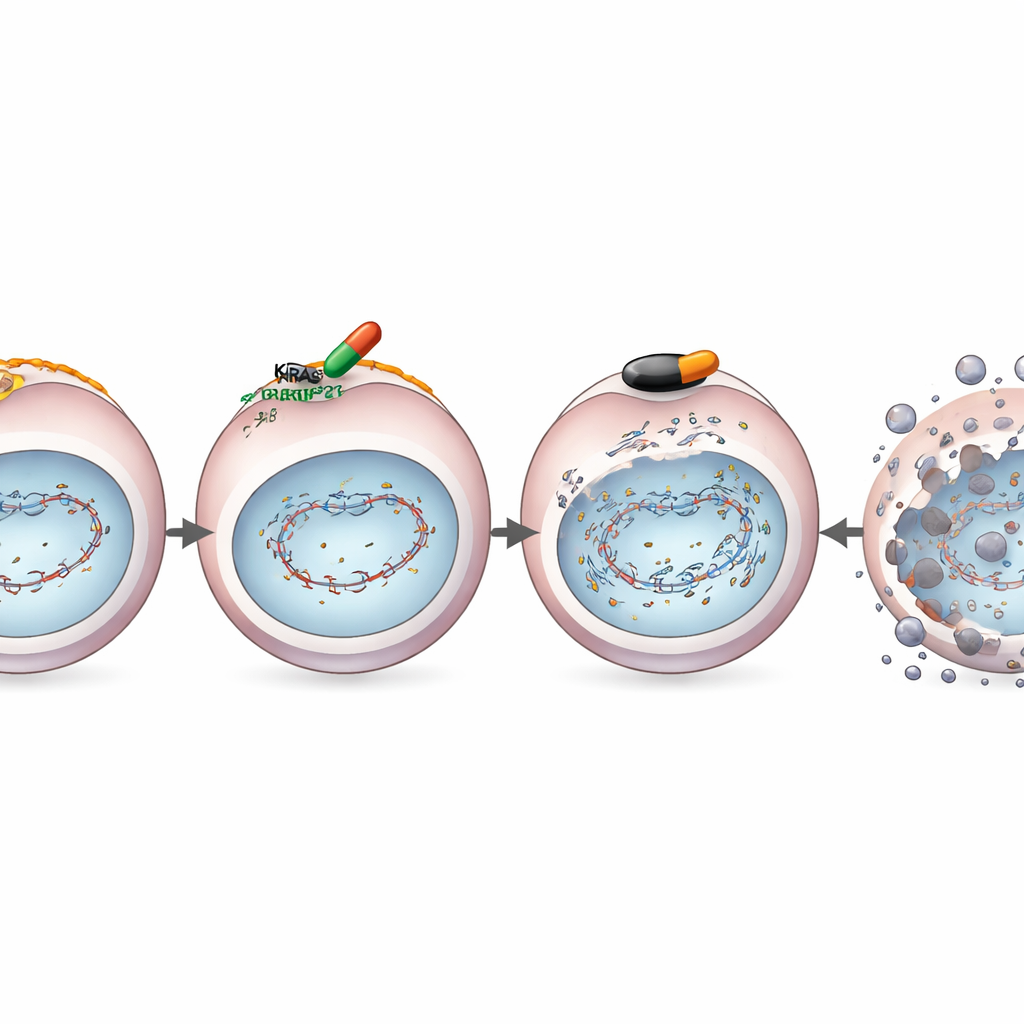
Turning a strength into a weakness
The team studied a highly selective KRASG12D-blocking drug called MRTX1133 in pancreatic cancer cells grown in the lab. When they treated KRASG12D-mutant cells with this drug and then exposed them to DNA-damaging radiation, the cells struggled to fix their broken DNA. Molecular tests showed why: MRTX1133 lowered the levels of key repair proteins, including BRCA1 and RAD51, which normally help patch up dangerous double-strand DNA breaks. Specialized reporter assays confirmed that the cells had become “homologous recombination deficient” — in plain terms, they lost one of their most accurate DNA repair systems.
Combining two targeted drugs for a stronger hit
Loss of this repair pathway is precisely the kind of flaw that makes cells vulnerable to a different class of drug called PARP inhibitors, already used in some breast and ovarian cancers. The researchers therefore combined MRTX1133 with the PARP inhibitor olaparib in KRASG12D-mutant pancreatic cancer cells and in mouse models. Across several cell lines, the drug pair worked together far better than either alone, killing more cancer cells and sharply reducing their ability to form new colonies. In mice carrying human or mouse pancreatic tumors with KRASG12D, the combination treatment shrank tumors more deeply and more durably than single drugs, and triggered more DNA damage and cancer cell death under the microscope, while sparing normal cells.
Working even when resistance appears
Targeted drugs like MRTX1133 often fail because tumors rewire their growth circuits and restore signaling through alternative routes. The team deliberately created cancer cell lines that had become resistant to MRTX1133’s growth-blocking effects. Strikingly, even in these resistant cells, the drug still turned down BRCA1, RAD51 and related repair proteins, keeping the DNA-repair weakness in place. As a result, pairing MRTX1133 with olaparib continued to show strong, cooperative killing of cancer cells in dishes and in mice bearing resistant tumors. This suggests that the combination attacks a fundamental vulnerability that persists even after classic resistance pathways switch back on.
Waking up the immune system
Beyond directly injuring tumor cells, the combined treatment also reshaped the tumor’s neighborhood. Using single-cell RNA sequencing and flow cytometry in immune-competent mice, the researchers found that combination therapy pulled more cancer-fighting CD8 and helper CD4 T cells into the tumors and pushed them into a more aggressive, “effector” state, while reducing signs of T-cell exhaustion. When CD8 T cells were experimentally removed, the benefit of the drug pair shrank, showing that immune attack is an important part of the overall effect. In other words, the strategy not only breaks the tumor from within by crippling DNA repair, it also invites the immune system to join the fight.
What this could mean for patients
Although the particular KRASG12D drug tested here is no longer moving forward clinically, the study delivers a clear message: selectively blocking KRASG12D can create a specific DNA-repair weakness that makes pancreatic tumors exquisitely sensitive to PARP inhibitors, and this holds true even after resistance to the KRAS drug itself arises. Future KRASG12D-targeted medicines could be paired with PARP inhibitors, and perhaps with immunotherapies, to turn a once “undruggable” mutation into a tailored treatment opportunity for the large fraction of pancreatic cancer patients whose tumors carry this genetic change.
Citation: Xu, X., Chen, X., Xu, R. et al. Combination of PARP and KRASG12D inhibitors enhances therapeutic efficacy by exploiting vulnerabilities in PDAC. Nat Commun 17, 3118 (2026). https://doi.org/10.1038/s41467-026-69695-4
Keywords: pancreatic cancer, KRASG12D, PARP inhibitor, DNA repair, combination therapy