Clear Sky Science · en
Exon inclusion signatures enable accurate estimation of splicing factor activity
Reading the Cell’s Hidden Editing Marks
Every cell in our body constantly edits its RNA messages before turning them into proteins. This editing, called splicing, helps decide whether a cell stays healthy or turns cancerous. The study behind this article shows that by looking carefully at which pieces of RNA are kept or skipped—called exon inclusion signatures—scientists can accurately infer the activity of the molecular "editors" that control splicing, even in complex diseases like cancer.
How Cells Cut and Paste Their Messages
Genes are not read in one continuous stretch. Instead, cells remove non‑coding segments and stitch together coding pieces, known as exons, to build final RNA messages. Specialized proteins called splicing factors guide this cut‑and‑paste process, deciding which exons are included. Their behavior is influenced by many layers of regulation: how much of their own RNA and protein is made, how they are chemically modified, where they sit inside the cell, and how they interact with other proteins. Because so many levers can change splicing factor behavior, simply measuring one type of data—such as gene expression—often fails to reveal what these factors are really doing.
Turning Exon Patterns into Activity Readouts
Inspired by earlier work on transcription factors, the authors propose a different strategy: instead of trying to measure splicing factors directly, read their activity from their effects. When a splicing factor changes, the inclusion of its target exons shifts in recognizable patterns. The team compiled hundreds of experiments in which individual splicing factors were knocked down, knocked out, or overexpressed, and used these data to build "empirical networks" linking each factor to the exons it clearly affects. They then adapted a computational framework called VIPER to read a new exon inclusion signature and score how active each splicing factor must be to explain the observed pattern.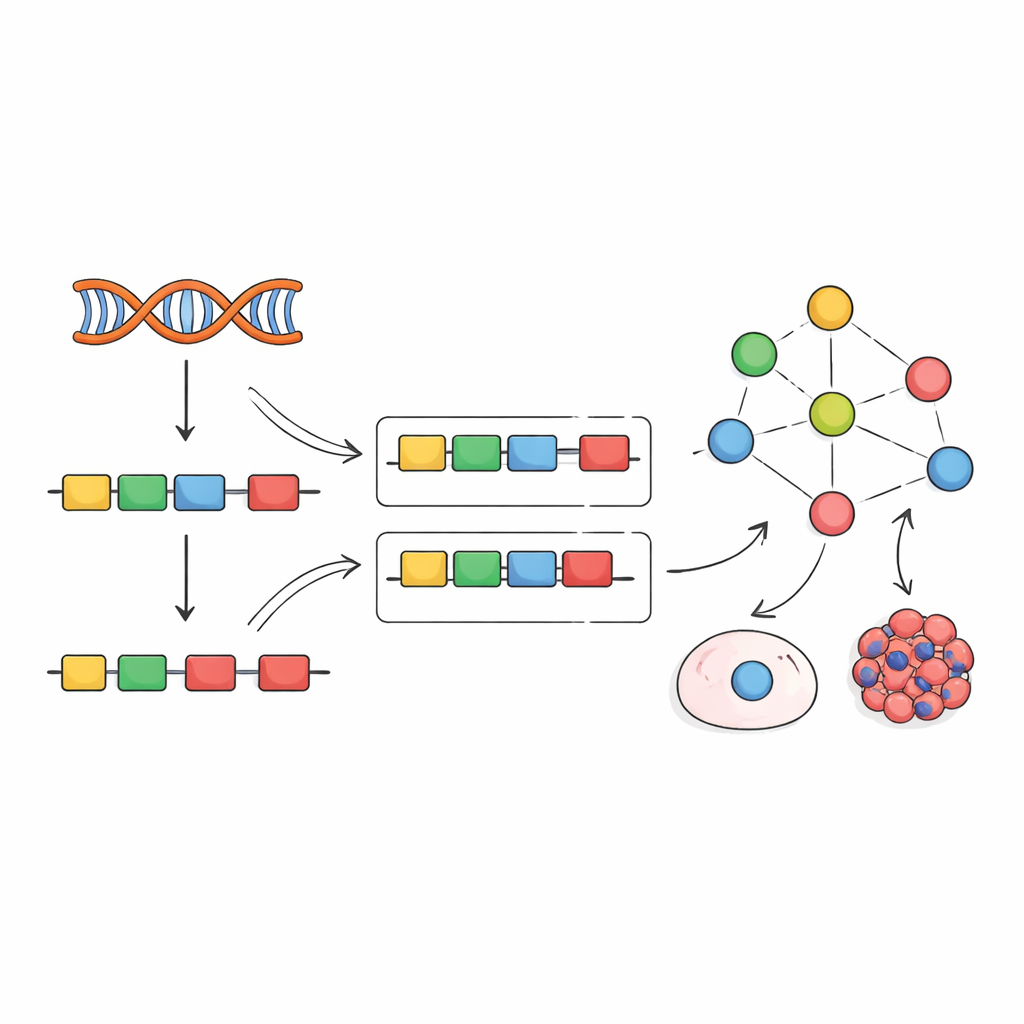
Testing the Method in Real‑World Perturbations
To see whether this approach works, the researchers benchmarked several ways to build networks and compute activity scores. Empirical networks derived directly from perturbation experiments, combined with VIPER’s enrichment analysis, clearly outperformed alternatives based on statistical inference alone. The method correctly identified the experimentally perturbed splicing factor in most tests, even across different cell types and studies. It also captured more subtle regulatory mechanisms. For example, a cancer drug called Indisulam triggers degradation of the splicing factor RBM39 protein while its RNA levels rise in what seems to be a compensation attempt. Traditional expression analysis would misleadingly suggest that RBM39 is more active, but the exon‑based activity score correctly revealed a strong loss of function, matching the known drug action.
Uncovering Hidden Splicing Programs in Cancer
Armed with this tool, the authors turned to The Cancer Genome Atlas, analyzing exon‑level data from multiple tumor types and matching healthy tissues. They discovered two broad and recurring splicing programs. One program consists of splicing factors that tend to be more active in tumors and is associated with poorer patient survival—an oncogenic‑like program. The other features factors systematically less active in tumors and linked to better outcomes, resembling tumor suppressors. These programs touch genes involved in fundamental cancer hallmarks such as rapid cell division and the ability of tumors to hide from the immune system. For instance, some exons regulated by the tumor‑suppressor‑like program appear to influence how well patients respond to immune checkpoint therapies, pointing to new markers or intervention points.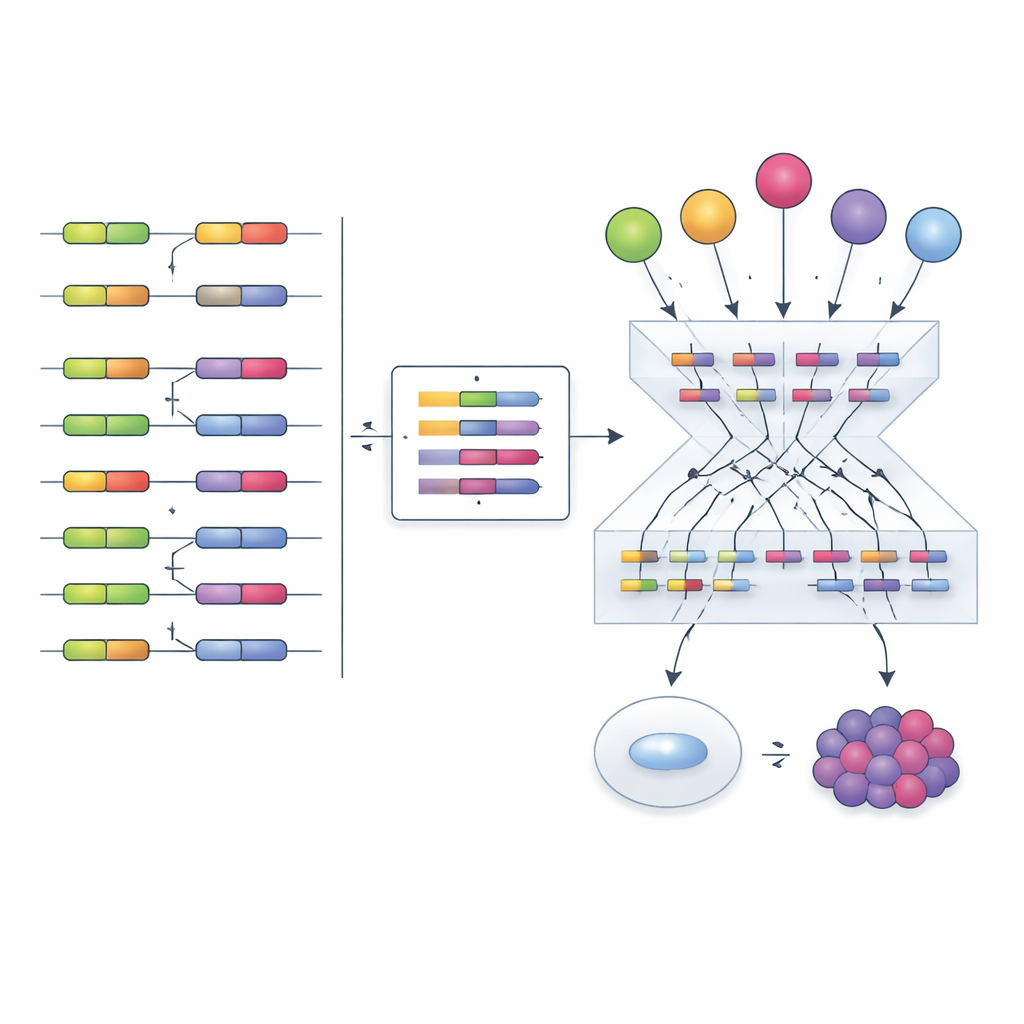
Following Splicing Changes Along the Road to Cancer
The team also examined a step‑by‑step model of human cells progressing from normal to immortalized, tumor‑forming, and finally metastasizing states. They found that the oncogenic‑like splicing program becomes more active as cells acquire cancer‑driving mutations, while the tumor‑suppressor‑like program fades. By integrating multiple data layers—RNA levels, protein abundance, chemical modifications, and splicing changes inside splicing factors themselves—they identified a focused set of candidate molecular events that may drive these program shifts, offering a prioritized list for future experimental testing.
Why This Matters for Patients and Future Research
In essence, the study shows that the complex behavior of splicing factors can be distilled into a single, interpretable activity score derived from how exons are included or skipped. This makes it possible to study splicing regulation in large patient cohorts and diverse experiments using only standard RNA sequencing data, without requiring expensive multi‑omic profiling. For a lay reader, the key message is that patterns in how genes are cut and pasted carry rich information about the hidden control systems of the cell, and that decoding these patterns can reveal new cancer drivers, improve prognosis, and guide the search for more precise treatments.
Citation: Anglada-Girotto, M., Segura-Morales, C., Moakley, D.F. et al. Exon inclusion signatures enable accurate estimation of splicing factor activity. Nat Commun 17, 1994 (2026). https://doi.org/10.1038/s41467-026-69642-3
Keywords: RNA splicing, splicing factors, cancer genomics, transcriptomics, protein activity inference