Clear Sky Science · en
Excess FGFR3 signaling in achondroplasia disrupts turnover of resting zone chondrocytes via CREB signaling
Why this bone growth study matters
Achondroplasia is the most common cause of genetic short-limb dwarfism. It affects not only height but also spinal health, mobility, and quality of life. Current treatments help but do not fully restore bone growth. This study uses a sophisticated mouse model to uncover a previously hidden problem zone in growing bones and points to a new signaling switch, called CREB, as a promising target for future therapies.
How healthy bones grow in length
Long bones, such as the femur in the thigh, lengthen at growth plates near their ends. These growth plates are organized into three main layers of cartilage cells. At the top sits the resting zone, where cells behave like a stem-cell pool, dividing slowly and sending daughter cells downward. Below that, the proliferative zone contains rapidly dividing cells stacked in orderly columns that drive lengthening. Furthest along, the hypertrophic zone holds enlarged, mature cells that help guide the formation of new bone. The balance between these zones keeps bones growing at the right rate and in the right shape.
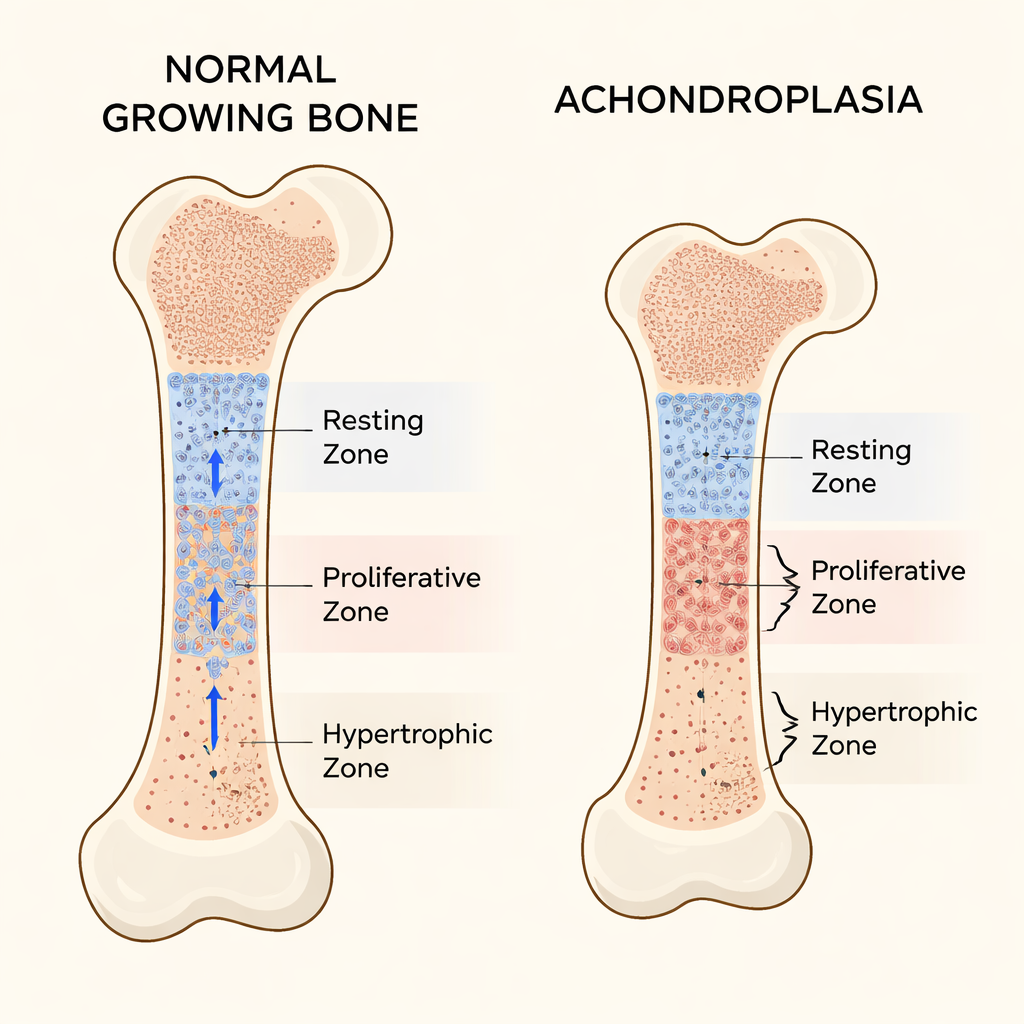
What goes wrong in achondroplasia
In most people with achondroplasia, a single mutation in the FGFR3 gene makes its receptor overactive, putting the brakes on bone growth. Prior work showed that this signaling slows down cell division in the proliferative zone and blocks the final enlargement of cells in the hypertrophic zone. Using mice engineered to carry the human achondroplasia mutation, the authors confirmed severe short limbs and shortened growth plates. But careful measurements revealed something that had been largely overlooked: the resting zone itself became unusually thick. Instead of acting as a steady, well-behaved stem-cell reservoir, this region expanded and contained cells poor in the normal cartilage matrix.
Resting zone cells lose their "stem-like" behavior
To understand this expansion, the team tracked how growth-plate cells divided and moved over time. In normal mice, resting zone cells divided rarely, and their descendants migrated downward in straight columns to refill the proliferative zone. In mutant mice, many more cells in the resting zone divided slowly and stayed put, building up a crowded layer that did not feed the lower zones properly. Lineage tracing with multicolor genetic labels showed that clonal columns were short and disorganized, with daughter cells wandering in random directions instead of forming neat stacks. Markers of stem-cell-like identity, such as the protein CD73, were lost in the expanded resting zone, suggesting that overactive FGFR3 had corrupted the normal stem-cell niche.
A new signaling culprit: CREB
The researchers then used single-cell RNA sequencing to profile thousands of individual growth-plate cells. They identified a distinct cluster that matched the expanded resting zone and was rich in a gene called Spon1, among others. Pathway analysis highlighted activation of CREB, a protein that turns genes on when it is switched into its phosphorylated form. Microscopy showed that resting zone cells in the mutant mice strongly expressed activated CREB and its co-activator CBP, along with high levels of FGFR3 and downstream molecules like STAT5. In cell culture, stimulating the FGFR3 pathway boosted CREB activity and increased SPONDIN1 (the SPON1 protein), while blocking FGFR3 or CREB reduced these signals. This placed CREB as a key relay between the overactive receptor at the cell surface and the faulty behavior of resting zone cells.
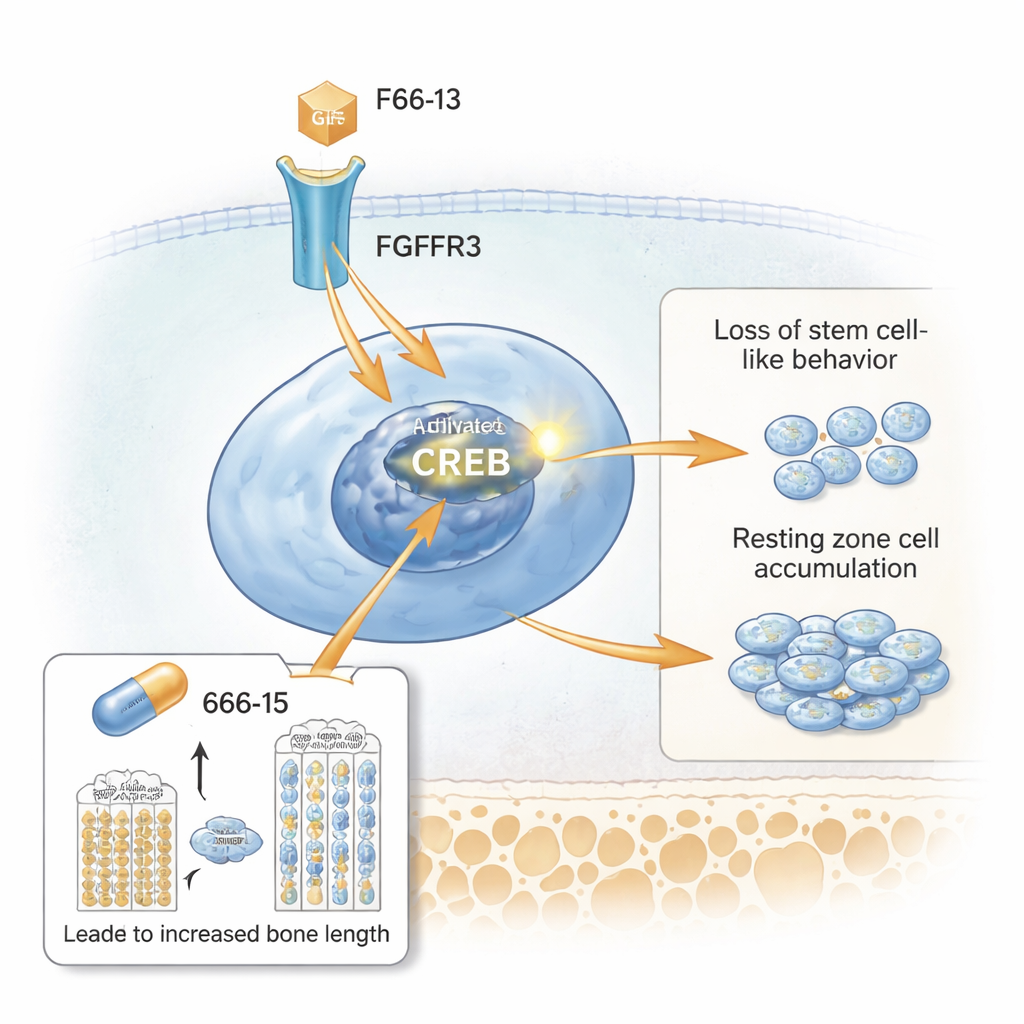
Testing a drug that tamps down CREB
Finally, the team asked whether turning down CREB could ease the growth problems. They treated achondroplasia-model mice with a small-molecule CREB inhibitor, called 666-15, during the rapid growth period after birth. Compared with untreated mutant mice, those given 666-15 had greater body weight and longer femurs. Their growth plates looked more normal: the resting zone thinned, the proliferative and hypertrophic zones recovered in height, and cartilage matrix proteins reappeared. Markers of overactive CREB signaling, including phospho-CREB, SPONDIN1, and STAT5, fell in the resting zone, while the stem-cell-like marker CD73 came back. Importantly, the same drug had little effect on healthy control mice at the tested dose, hinting that it mainly acts when CREB is abnormally high.
What this means for future treatments
The study shows that in achondroplasia, overactive FGFR3 does more than slow dividing and enlarging cells; it also derails the quiet, stem-cell-like resting zone by switching on CREB. This disruption starves the lower growth-plate layers of new cells and contributes to short bones. Existing drugs like vosoritide mainly target other pathways in the proliferative and hypertrophic zones and only partially restore bone length. By adding CREB to the list of targets—especially in the resting zone—future combination therapies might more fully normalize growth in children with achondroplasia.
Citation: Horike, N., Oura, S., Koyamatsu, S. et al. Excess FGFR3 signaling in achondroplasia disrupts turnover of resting zone chondrocytes via CREB signaling. Nat Commun 17, 1856 (2026). https://doi.org/10.1038/s41467-026-69507-9
Keywords: achondroplasia, FGFR3, growth plate, cartilage stem cells, CREB signaling