Clear Sky Science · en
High-throughput chemical proteomics workflow for profiling protein citrullination dynamics
Why changing protein building blocks matters
Our cells constantly fine‑tune how proteins behave by adding small chemical tweaks after they are made. One such tweak, called citrullination, subtly changes the charge of a common amino acid and can reshape how proteins fold, stick to DNA, or interact with other molecules. These tiny edits are increasingly linked to autoimmune diseases, infections, cancer, and brain function—but they are notoriously hard to see. This study introduces a high‑throughput laboratory workflow that finally makes it possible to map citrullination across thousands of proteins, revealing when and where it appears in tissues and immune cells.
A hidden switch on proteins
Citrullination occurs when enzymes called PADs chemically modify the amino acid arginine, removing its positive charge. That seemingly small change can loosen how DNA is packed, alter the stiffness of structural proteins, or change how the immune system recognizes our own tissues. Abnormal citrullination has been implicated in rheumatoid arthritis, neurodegenerative disorders, viral infections, and cancer. Yet citrullinated proteins are rare and easily confused with other, more common modifications, so standard mass spectrometry methods often miss them. The result is that researchers have had only a fragmented view of the body’s “citrullinome”—the full set of citrullinated proteins.
A two‑step tag‑and‑catch strategy
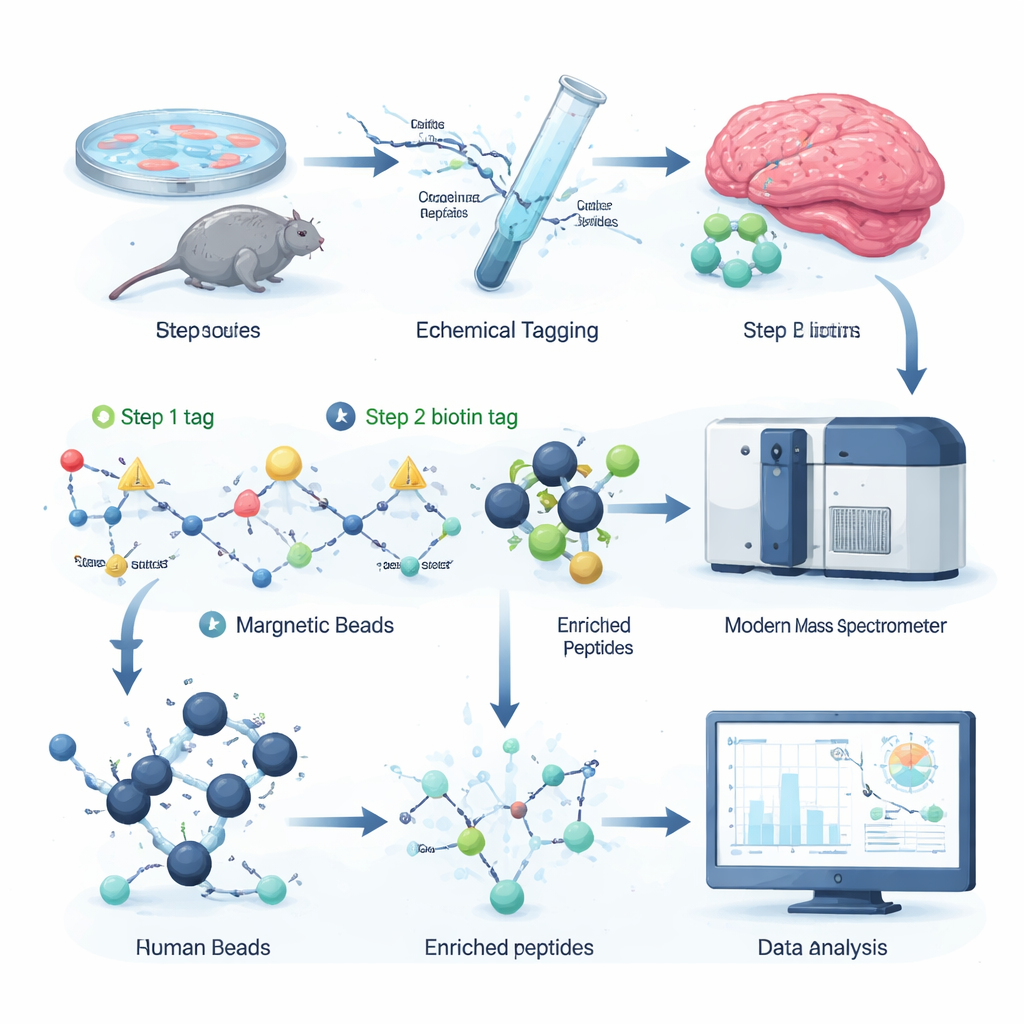
The authors devised a chemical labeling strategy that selectively attaches a removable “handle” to citrullinated sites on peptides, the fragments of proteins analyzed by mass spectrometry. In the first step, a small reactive molecule recognizes citrulline and installs a tiny tag. In the second, a larger biotin tag is clicked on, allowing the labeled peptides to be grabbed out of a complex mixture with streptavidin beads, a common biochemical tool. A gentle chemical treatment then snips off the bulky part of the tag, leaving behind a small, well‑defined mass shift that the mass spectrometer can easily detect. Because all reagents are commercially available and the entire protocol fits into 96‑well plates, the workflow is fast, scalable, and compatible with existing proteomics setups.
Seeing more of the citrullinome
By spiking known citrullinated peptides into cell extracts and systematically diluting them, the team showed that their enrichment strategy boosts the signal of citrullinated peptides more than tenfold, even when they represent less than one in a thousand molecules. In complex samples, the number of citrullinated sites detected and their measured intensities jumped dramatically after enrichment. Applying the method to mouse brain tissue revealed two to three times more distinct citrullination sites than a previous state‑of‑the‑art approach, including many on myelin basic protein, which insulates nerve fibers, and on proteins involved in synaptic communication. This suggests that citrullination may influence both how nerve cells signal and how brain wiring is maintained.
Immune cells casting sticky protein webs
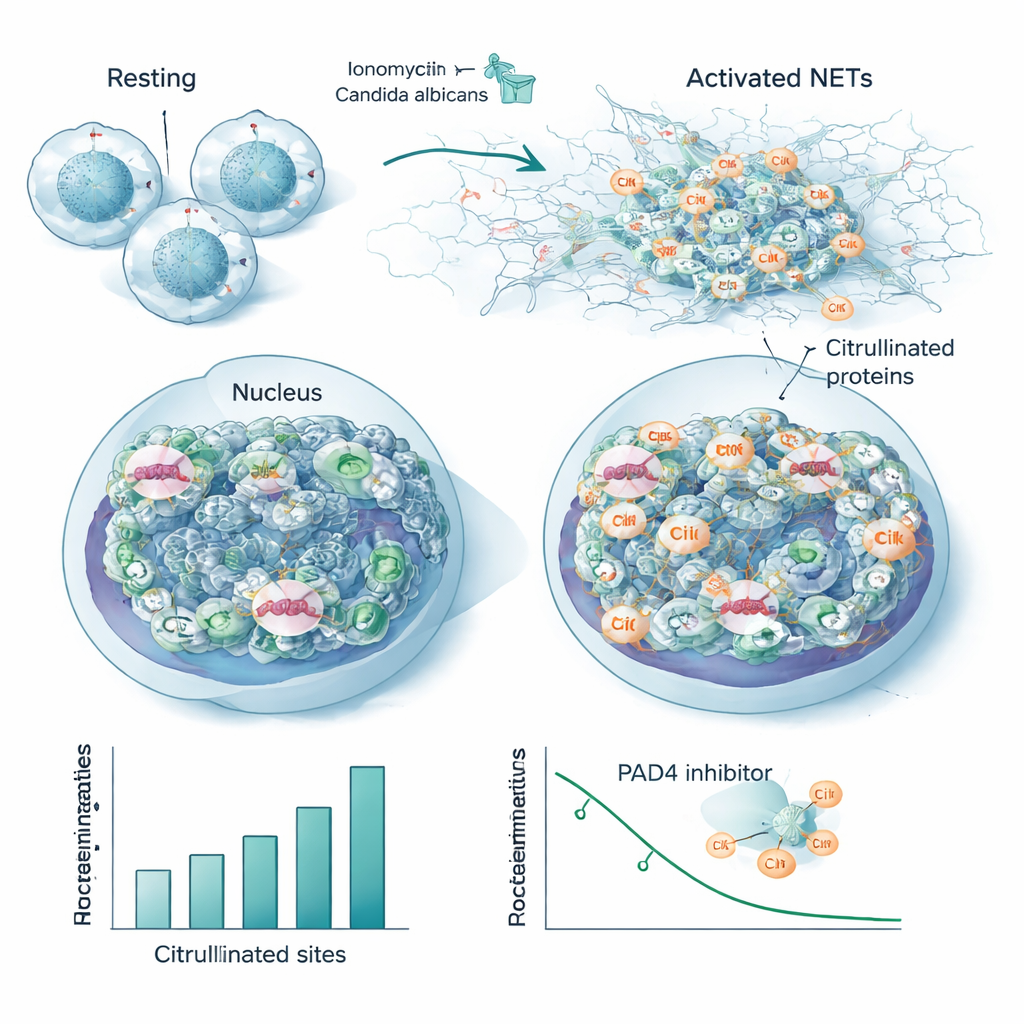
Neutrophils, a frontline white blood cell type, can fight invaders by releasing sticky webs of DNA and proteins called neutrophil extracellular traps, or NETs. NET formation depends on PAD4, a citrullinating enzyme that loosens chromatin so DNA can spill out. Using their new workflow, the researchers followed how citrullination changed in human neutrophils exposed to increasing doses of a chemical activator. They detected up to 1,700 citrullinated peptide fragments across 580 proteins, with hundreds of sites rising or falling in a dose‑dependent manner while overall protein levels stayed constant. Histones—the proteins that package DNA—showed widespread citrullination, not just at a few classic sites, and linker histone H1 variants were especially modified. Structural proteins such as actin regulators and lamin B, which shapes the nuclear envelope, also became heavily citrullinated, pointing to a coordinated softening of both chromatin and cellular scaffolds during NET release.
A core citrullination signature in infection
To mimic a real infection, the team stimulated neutrophils with heat‑killed Candida albicans, a common fungal pathogen. Although this produced fewer modified sites overall than the strong chemical activator, the vast majority of citrullinated proteins and positions overlapped between the two triggers. This overlap defines a conserved “core citrullinome” associated with NET formation, including many nuclear and cytoskeletal proteins and several known autoantigens—the very targets of antibodies in autoimmune diseases. When the researchers added a PAD4‑blocking drug, many of these same sites lost their citrullination in a dose‑dependent fashion, directly tying them to the enzyme’s activity and suggesting they could serve as sensitive readouts of PAD4 inhibition.
What this means for health and disease
By turning an elusive modification into a measurable signal, this workflow makes it possible to chart where and when citrullination occurs in tissues, immune responses, and disease models. For non‑specialists, the key message is that citrullination acts like a subtle molecular dimmer switch on proteins, and being able to see its patterns in high detail could help explain how autoimmune diseases start, how infections reshape immune cells, and how brain proteins change over time. The method’s scalability and reliance on standard lab equipment mean it can be widely adopted, opening the door to discovering new drug targets, precision diagnostics, and a deeper understanding of how small chemical edits can have large biological consequences.
Citation: Meelker González, R., Laposchan, S., Riedel, E. et al. High-throughput chemical proteomics workflow for profiling protein citrullination dynamics. Nat Commun 17, 1982 (2026). https://doi.org/10.1038/s41467-026-69490-1
Keywords: citrullination, autoimmune disease, neutrophil extracellular traps, mass spectrometry, post-translational modification