Clear Sky Science · en
Pathophysiological significance of impaired KAT7-dependent histone H3K14 acetylation during zinc deficiency
Why tiny nutrients matter for our health
Zinc is a trace metal that our bodies need only in pinches, yet it quietly supports hundreds of proteins that keep cells running. When zinc is lacking—because of diet, illness, or aging—it has been linked to problems ranging from poor growth to weakened immunity and fatty liver disease. This study asks a deeper question: how do cells actually sense that zinc is running low, and how can that shortage be translated into long‑lasting changes in gene activity and organ health?
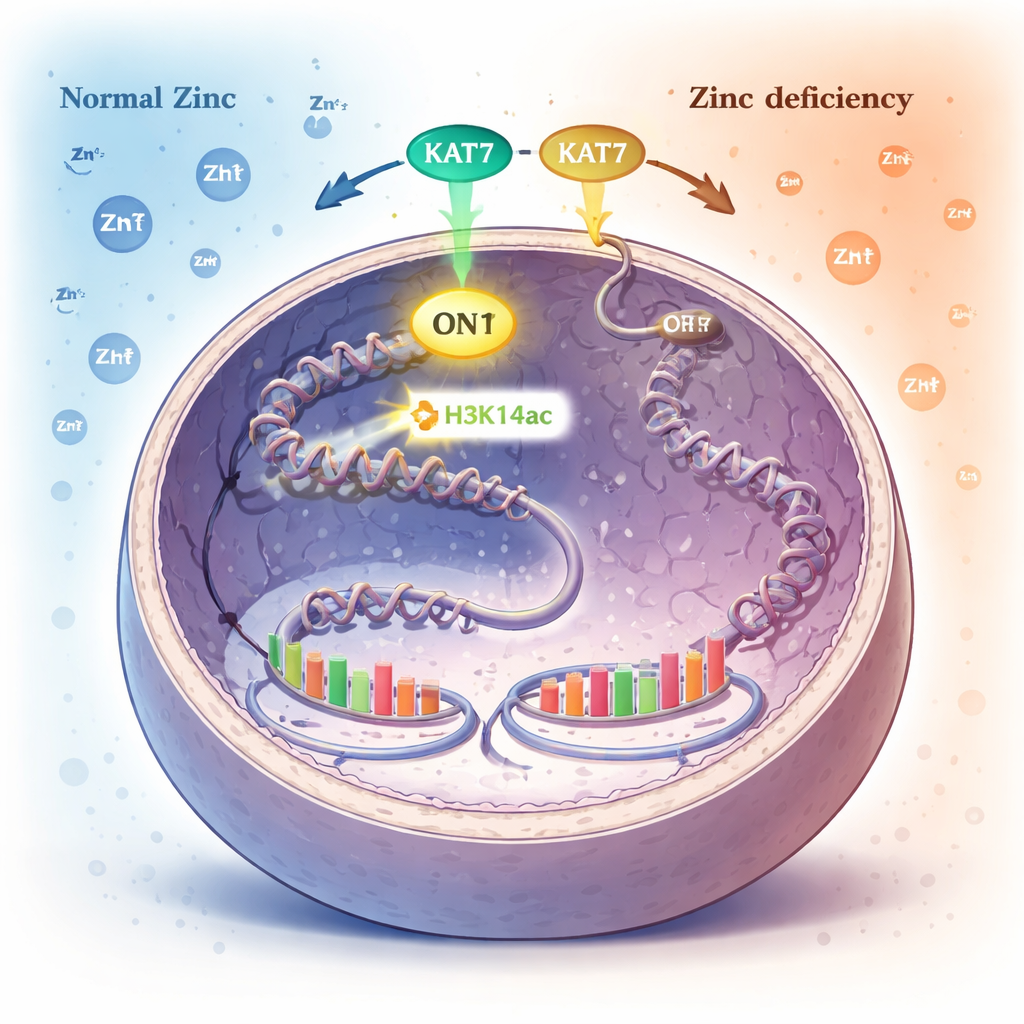
A chemical tag on DNA packaging as an internal zinc alarm
Inside the nucleus, DNA is wrapped around protein spools called histones. Cells control which genes are active by adding or removing tiny chemical tags to these histones. One such tag, called acetylation at a specific spot on histone H3 (H3K14ac), is added by an enzyme named KAT7. The authors discovered that when zinc becomes scarce, levels of this H3K14ac tag fall dramatically, while many other common histone tags stay unchanged. This pointed to H3K14ac, and the KAT7 enzyme that creates it, as a key sensor of zinc status.
How zinc keeps a key enzyme switched on
By systematically disabling different enzymes, the researchers showed that KAT7 is the main source of H3K14ac in human cells. KAT7 contains a small zinc‑binding structure within its active center. When cells were pushed into zinc deficiency, KAT7’s ability to place the H3K14ac mark declined, even though the protein itself remained in the nucleus and stayed associated with its helper partners. Detailed tests with purified KAT7 fragments revealed that properly bound zinc in this region is essential for its activity; disrupting zinc binding shut the enzyme down, and carefully adding zinc back restored function. In essence, KAT7 behaves like a zinc‑dependent switch that controls a specific histone mark.
Turning zinc loss into gene changes that rescue zinc levels
What does the loss of this histone mark actually do? Using genome‑wide mapping, the team showed that H3K14ac is especially enriched at enhancer regions—regulatory stretches of DNA that fine‑tune nearby genes. Under zinc‑deficient conditions, H3K14ac was stripped from many enhancers, and the greater the loss, the stronger the change in neighboring gene activity. One standout gene was ZIP10, which encodes a protein on the cell surface that imports zinc. When H3K14ac fell at ZIP10’s enhancer, ZIP10 levels on the membrane rose, allowing more zinc to flow into the cell. Blocking KAT7 or preventing H3K14ac loss interfered with this response and reduced zinc uptake, even after zinc was added back. This shows that cells convert zinc scarcity into an epigenetic signal that boosts zinc‑import machinery to restore balance.
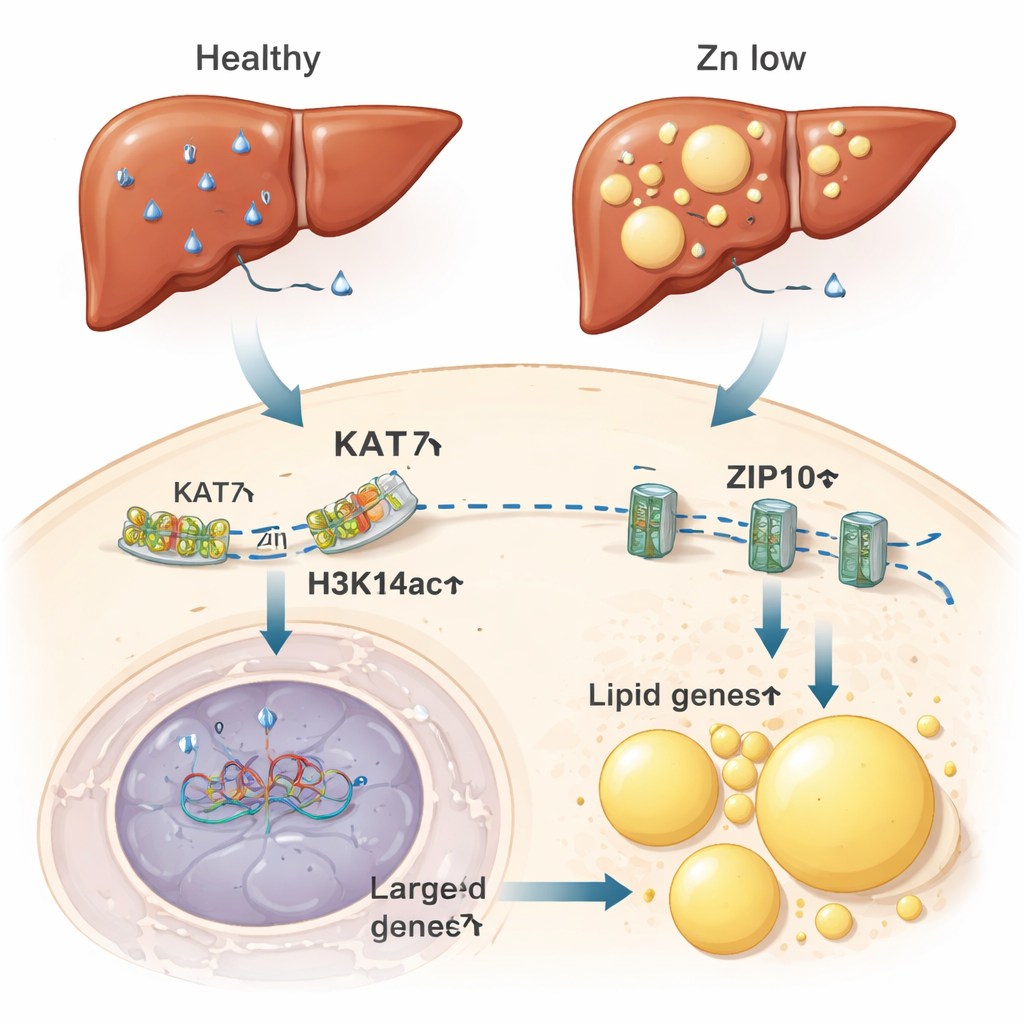
From zinc‑starved cells to fatty livers
The authors next asked whether this zinc‑sensitive switch has consequences in whole animals. In mice fed a zinc‑deficient diet, the liver—a central hub of zinc and fat metabolism—showed reduced zinc levels, lower H3K14ac, and weakened KAT7 activity. These changes coincided with higher expression of genes that drive fat storage and the formation of lipid droplets, the microscopic fat packets inside cells. The livers of zinc‑deficient mice accumulated fat to a degree comparable to those on a high‑fat diet. Remarkably, simply lowering KAT7 activity with a drug, even without changing dietary zinc, was enough to promote fat build‑up in liver cells. Conversely, supplying extra zinc lessened fat accumulation caused by a high‑fat diet.
What this means for human disease risk
Putting their findings in a clinical context, the researchers reviewed human studies that measured zinc levels in liver tissue. Across multiple reports, people with fatty liver and related disorders had significantly less zinc in their livers than healthy controls. Together with the mouse experiments, this suggests that chronic zinc deficiency may promote fatty liver disease by silencing KAT7, erasing the H3K14ac mark, and persistently turning up genes that favor fat storage. In simple terms, the work reveals an internal “zinc‑to‑epigenetics” circuit: when zinc falls, a zinc‑dependent enzyme loses power, altering DNA packaging in ways that first help cells pull in more zinc, but over time can also push the liver toward unhealthy fat accumulation.
Citation: Fujisawa, T., Takenaka, S., Maekawa, L. et al. Pathophysiological significance of impaired KAT7-dependent histone H3K14 acetylation during zinc deficiency. Nat Commun 17, 1710 (2026). https://doi.org/10.1038/s41467-026-69476-z
Keywords: zinc deficiency, epigenetics, liver fat, histone acetylation, zinc transporters