Clear Sky Science · en
A generic reference defined by consensus peaks for single-cell ATAC-seq data analysis
Why mapping our DNA’s open doors matters
Every cell in your body carries essentially the same DNA, yet brain cells, blood cells, and tumor cells behave very differently. One key reason is that only certain stretches of DNA are exposed and "open" for use at any given time. New single-cell technologies can now measure this openness genome-wide, but until now they have lacked a common reference map—something like a standard atlas—to compare results across experiments and labs. This study builds such a map, called cPeaks, and shows how it can sharpen our view of cell types, development, and cancer.
Turning many experiments into one shared map
The authors started by gathering 624 high-quality experiments that measured open chromatin—the accessible parts of DNA—across more than 40 human organs. In each experiment, computer programs had already marked "peaks" where the DNA was especially exposed. Instead of treating each dataset separately, the team carefully overlaid all of these peak lists along the genome and merged overlapping regions. They then examined how often each tiny position inside these merged regions was called open across experiments, turning each region into a characteristic shape that reflected how consistently it appeared. When a merged region actually contained several closely spaced open sites, they split it into multiple simpler units. These units—about 1.4 million in total—became the observed consensus peaks, or cPeaks, a candidate reference catalog for human chromatin accessibility. 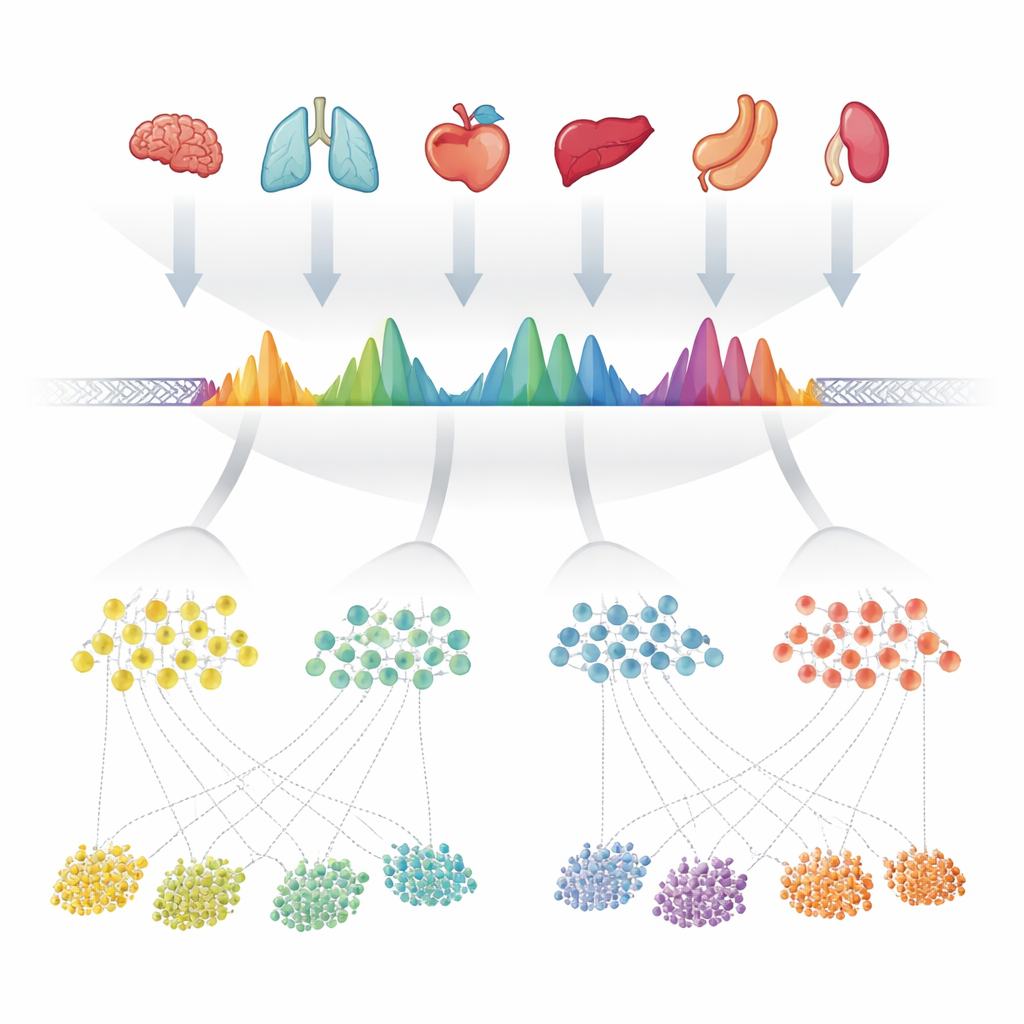
A stable fingerprint across tissues and technologies
To be a useful reference, these cPeaks must represent genuine, repeatable features of the genome, not quirks of particular samples or software. The authors tested this by recreating their merged regions using only blood samples, only solid tissues, separate public databases, and even different laboratory methods for probing open DNA. In each case, the same genomic locations produced remarkably similar peak shapes, and most single-cell datasets they examined overlapped more than 90% of their own peaks with the cPeak catalog. Reads from many organs piled up precisely around cPeak centers, showing that these regions reliably capture where chromatin is open. Compared with previous reference sets based on related technologies, cPeaks covered more of the accessible DNA picked up by ATAC-seq experiments, and captured nearly as much signal as peaks defined fresh in each dataset—despite being fixed and reusable.
Teaching a neural network to find missing regions
Even hundreds of existing samples cannot cover every possible cell type. To extend their map into regions that had not yet been observed, the team turned to deep learning. They trained a one-dimensional convolutional neural network on DNA sequences: examples that lay within observed cPeaks served as positives, while randomly chosen background regions served as negatives. The model learned to distinguish these two with high accuracy, implying that cPeaks carry recognizable sequence patterns. When the researchers deliberately hid peaks specific to one tissue at a time, the network still recovered them from sequence alone, including rare, tissue-specific sites. They then slid a small window across the rest of the genome, scoring each segment and adding about 280,000 high-scoring new regions to the catalog as predicted cPeaks, particularly improving coverage in tissues underrepresented in the original data.
Linking open regions to genes, cell types, and rare cells
With a richer reference in hand, the authors asked what these regions do. Many cPeaks lie near gene start and end sites or overlap known regulatory elements such as promoters, enhancers, and binding sites for architectural proteins like CTCF. A small subset is accessible in almost every dataset; these longer "housekeeping" cPeaks tend to sit in core promoter regions of genes needed for basic cell maintenance. The team also classified cPeaks by how sharp and consistent their edges are across samples, which reflects how precisely nearby DNA is packaged into nucleosomes. Regions with sharply defined boundaries are enriched for particular families of transcription factors that are known to reshape chromatin and drive development. When cPeaks were used as the feature set to analyze multiple single-cell datasets, they improved the accuracy of cell type labeling, and were especially helpful in identifying rare cell types and subtle subtypes that previous peak sets or simple genomic grids often blurred together.
Following development and cancer using a common language
The power of a standard reference becomes clear when comparing very different biological contexts. Using cPeaks, the authors reanalyzed single-cell data from developing human retina, large atlases of fetal and adult tissues, and several cancers. They could reconstruct developmental trajectories and see that the fraction of sharply bounded, "well-positioned" cPeaks tends to rise during transitional stages, then fall as cells settle into stable identities. A similar pattern appeared across tumor stages: intermediate cancers showed a higher proportion of these structured regions, hinting at intense regulatory remodeling. In one ovarian tumor, cPeaks helped reveal two distinct cancer cell subclones with different DNA copy-number changes, showcasing how the reference can expose hidden complexity in disease.
What this means for future genome research
For non-specialists, cPeaks can be thought of as a standardized set of coordinates marking where the genome is most likely to be physically open and active across many human cell types. By aligning new single-cell chromatin experiments to this shared map, researchers can compare results across studies, more easily spot rare or transitional cell states, and begin to build large-scale models of gene regulation—much as standardized gene catalogs enabled the rise of single-cell RNA atlases. The current cPeak catalog is a first draft that will grow as new data arrive, but it already provides a common language for describing chromatin accessibility, bringing us closer to a unified view of how DNA packaging guides development, health, and disease. 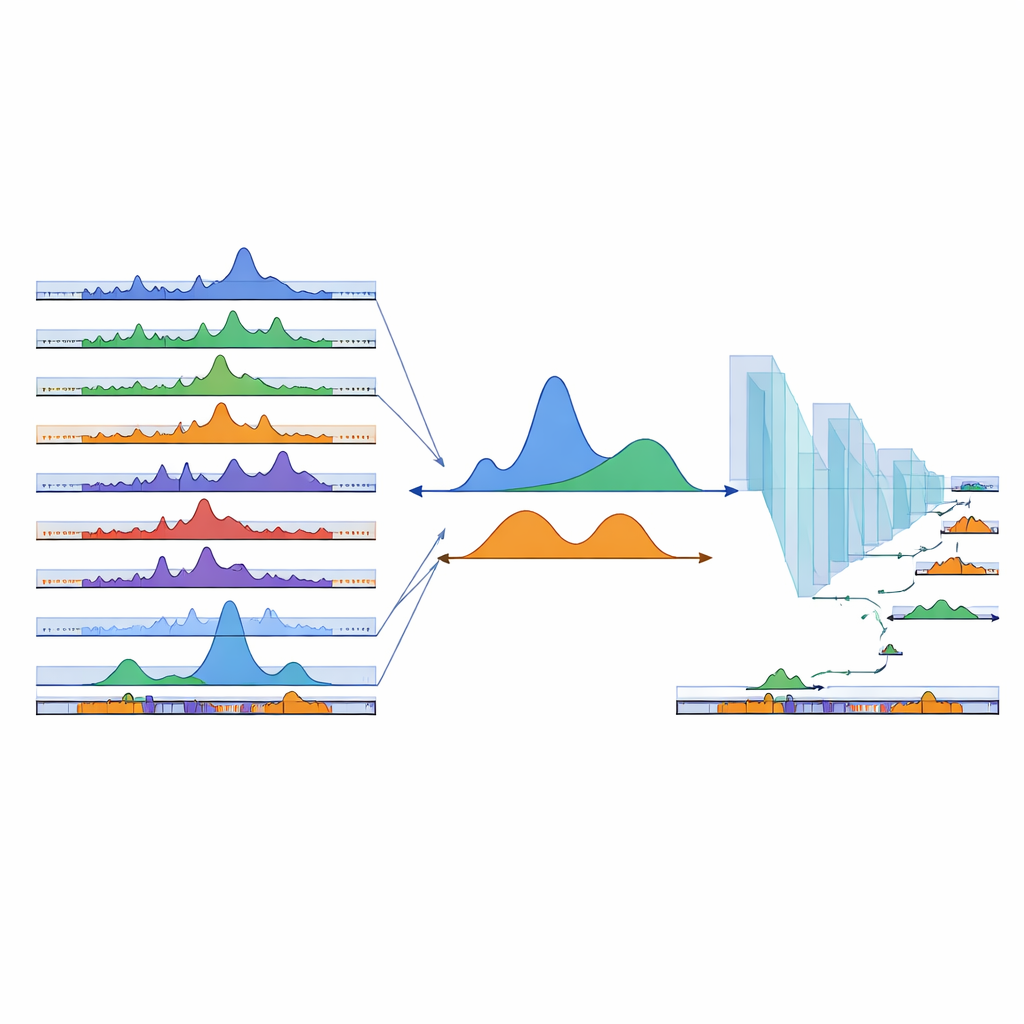
Citation: Meng, Q., Wu, X., Chen, W. et al. A generic reference defined by consensus peaks for single-cell ATAC-seq data analysis. Nat Commun 17, 2522 (2026). https://doi.org/10.1038/s41467-026-69461-6
Keywords: chromatin accessibility, single-cell ATAC-seq, consensus peaks, gene regulation, deep learning genomics