Clear Sky Science · en
Triple targeting of STING, TGF-β, and PD-L1 boosts CXCL16–CXCR6 signaling for potent antitumor response
Turning Cold Tumors Hot
Cancer immunotherapy has transformed treatment for some patients, yet many tumors still shrug off these powerful drugs. This study explores why certain cancers resist modern immune “checkpoint” drugs and proposes a smarter, three-pronged attack that wakes up the body’s defenses, draws in elite killer T cells, and keeps them active right inside the tumor.
Why Current Immune Drugs Aren’t Enough
Most approved immunotherapies target a single brake on immune cells, such as the PD-1/PD-L1 pathway. A newer class of drugs tries to go further by also blocking TGF-β, a molecule that strongly suppresses immunity in advanced cancers. One such drug, YM101, combines TGF-β and PD-L1 blockade in a single antibody and showed promise in mice. But even in genetically identical animals, some tumors barely shrank. By comparing responsive and resistant tumors, the researchers found that successful treatment went hand in hand with strong “innate” immune activation, especially signaling through a pathway called STING, which senses abnormal DNA and triggers antiviral-like alarm signals.
Adding a Third Lever: The STING Pathway
Suspecting that weak innate activation was the missing piece, the team combined YM101 with a pill-like STING agonist called MSA-2 across several mouse tumor models, including typically hard-to-treat “cold” tumors. The triple approach—STING activation plus TGF-β and PD-L1 blockade—shrank tumors more effectively, extended survival, and often protected mice from tumor regrowth after re‑challenge, indicating long-lasting immune memory. This outperformed the more conventional pairing of a STING agonist with PD-L1 blockade alone, and even boosted STING agonist therapy when only TGF-β was blocked, revealing that TGF-β itself acts as a major brake on STING-driven immunity. 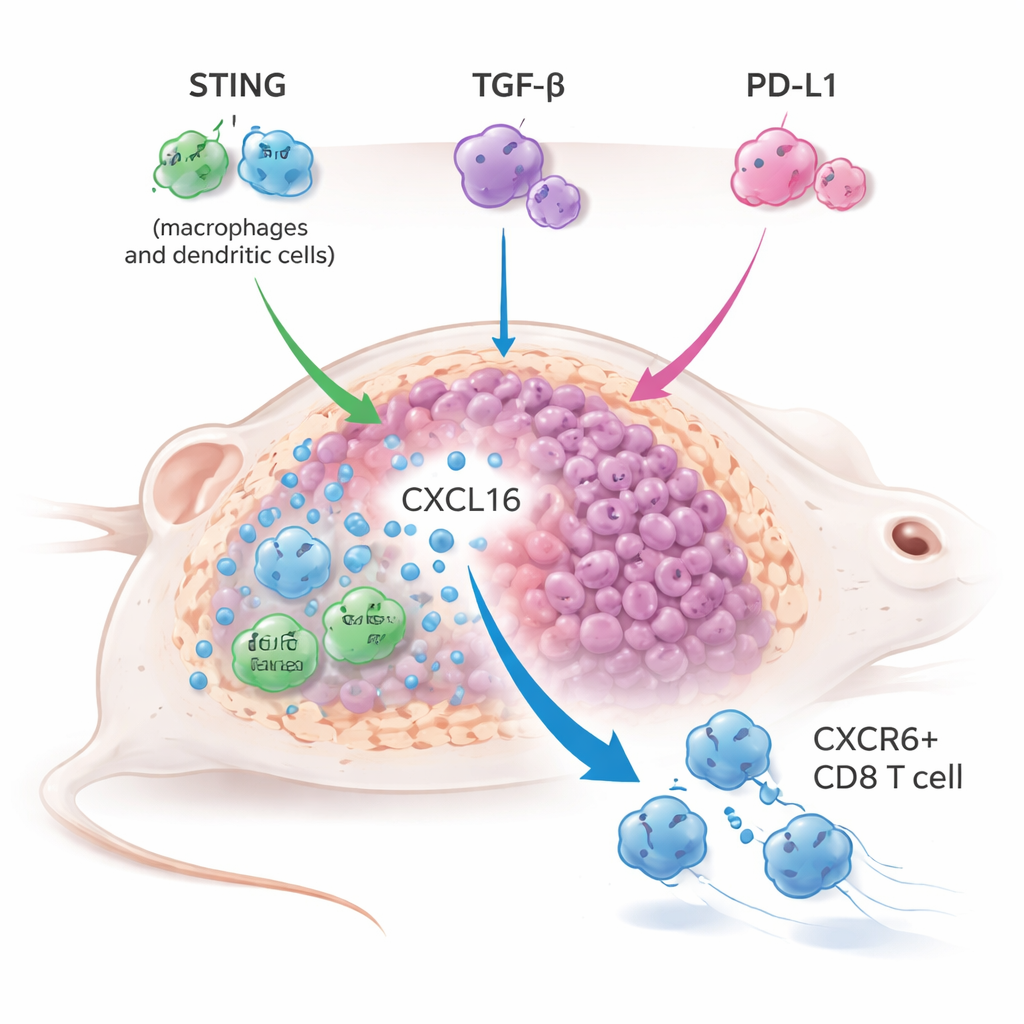
Recruiting a Specialist Killer Squad
To understand how this triple strategy worked, the researchers used single-cell RNA sequencing and detailed immune profiling of treated tumors. They discovered a striking expansion of a particular killer T cell subset marked by the receptor CXCR6. These CXCR6+ CD8 T cells were highly armed, expressing high levels of granzymes, perforin, and inflammatory molecules, and showed strong signs of activation and proliferation. At the same time, tumor-associated macrophages and dendritic cells ramped up production of a chemokine called CXCL16, which binds CXCR6 and helps retain these T cells in the tumor. When the CXCL16–CXCR6 link was broken—either by blocking CXCL16 or genetically deleting CXCR6 in T cells—the combined treatment largely lost its antitumor power, proving this axis is central to the therapy’s success.
How the Signal Cascade Is Switched On
Diving deeper, the team asked how STING activation and TGF-β blockade jointly boost CXCL16. In human and mouse immune cells, STING agonists strongly increased CXCL16 and the antiviral cytokine IFN‑β, while added TGF-β sharply reduced both. The researchers showed that STING triggers IFN‑I signaling, which activates the transcription factor STAT1; STAT1 then binds directly to the CXCL16 gene’s control region, turning it on. TGF-β disrupts this chain by interfering with a key step in STING signaling, likely via a protein called HDAC4 and reactive oxygen species, blunting IRF3 activation and downstream IFN‑β and CXCL16 production. Blocking TGF-β removes this brake, allowing STING agonists to fully ignite the STAT1–CXCL16 pathway in myeloid cells and thereby feed CXCR6+ T cells with the signals they need to stay and fight in the tumor.
Building a Single Precision Drug
To make this complex regimen more practical and tumor‑focused, the researchers engineered a single “immune-stimulating antibody conjugate” called Y101S. This molecule combines the dual TGF-β/PD-L1–blocking antibody with a STING agonist tethered by a cleavable linker. Y101S homes to PD-L1–positive myeloid cells in the tumor, is internalized, and then releases the STING drug inside these cells. In several mouse cancers, Y101S matched or exceeded the efficacy of giving YM101 plus a high-dose free STING agonist, despite carrying only a tiny fraction of that STING dose. It boosted CXCL16+ macrophages and dendritic cells, expanded CXCR6+ CD8 T cells, induced durable immune memory, and concentrated inflammatory signals in tumors while sparing healthy organs, with a favorable safety profile in mice. 
What This Means for Future Cancer Treatment
For non-specialists, the key message is that attacking cancer with just one or two immune switches may not be enough—especially when tumors actively silence early alarm systems. This work shows that combining STING activation with blockade of TGF-β and PD-L1 can rewire the tumor environment, powerfully attract and sustain a specialized group of killer T cells, and achieve deeper, more durable responses in preclinical models. The triple-targeting antibody–drug Y101S embodies this strategy in a single, targeted medicine and offers a roadmap for next-generation immunotherapies aimed at tumors that currently resist standard checkpoint drugs.
Citation: Yi, M., Li, T., Gu, Y. et al. Triple targeting of STING, TGF-β, and PD-L1 boosts CXCL16–CXCR6 signaling for potent antitumor response. Nat Commun 17, 1441 (2026). https://doi.org/10.1038/s41467-026-69456-3
Keywords: cancer immunotherapy, STING pathway, TGF-beta blockade, PD-L1 antibody, CXCL16 CXCR6 T cells