Clear Sky Science · en
Sequencing DNA methylation and hydroxymethylation at co-occurring chromatin features
Reading Our Cells’ Chemical Notes
Every cell in the body carries the same DNA, yet brain cells, skin cells, and stem cells behave very differently. One reason is that cells write chemical "notes" on DNA and its packaging proteins, helping to switch genes on or off. Until now, scientists have struggled to read several of these notes together on the very same stretch of DNA, leaving a gap in our understanding of how they work as a team. This study introduces a new way to read both the genetic code and key chemical markings at once, revealing how they combine to control important DNA switches called enhancers.
Why DNA Needs Pencil Marks
DNA does not act alone. It is wrapped around proteins called histones to form chromatin, and both DNA and histones can be decorated with small chemical groups. Two important marks on DNA are methyl and hydroxymethyl groups added to the letter C (cytosine). These marks influence how tightly DNA is packed and whether nearby genes are active. In broad strokes, methyl marks are often linked with gene silencing, while hydroxymethyl marks tend to appear where genes are active. But the impact of these marks depends on their local setting: exactly where they sit in the genome and which histone marks they sit next to.
The Trouble with Separate Maps
Existing sequencing methods can map methyl and hydroxymethyl marks across the entire genome, and other methods map histone marks that flag active or silent regions. However, these are usually done in separate experiments and then compared on a computer. That tells us which features tend to lie in the same neighborhood, but not whether they truly coexist on the very same piece of DNA in a single cell. Older attempts to combine these measurements relied on harsh chemical treatment that damages DNA and, crucially, could not reliably distinguish methyl from hydroxymethyl marks on the same read. As a result, researchers lacked a clear, molecular-level picture of how combinations of marks cooperate.
A New Multi-Layer Reading Method
The authors developed a method called 6-base-CUT&Tag that can read all four DNA letters plus two chemical states of cytosine—plain, methyl, and hydroxymethyl—on DNA fragments that are physically attached to chosen chromatin features. First, they use antibodies like molecular hooks to pull out DNA wrapped around histones carrying a specific mark, for example a tag of active chromatin. An engineered enzyme then tucks in special adapters, turning each captured DNA fragment into a small loop that survives cleanup steps that destroy stray pieces. A refined chemical and enzymatic process then converts the various cytosine states into distinct sequence signals, which modern sequencers can read. In this way, a single read reports where the fragment came from, what histone mark it carried, and which cytosines on it were methylated or hydroxymethylated.
Zooming In on Gene Switches
Using mouse embryonic stem cells as a test case, the team applied 6-base-CUT&Tag to several key histone marks that label different kinds of regulatory DNA. They focused on enhancers—stretches of DNA that act as switches to control when and where genes turn on. Enhancers can be in “active,” “primed,” or “poised” states, distinguished by particular histone marks. The researchers found that enhancers marked only by a histone tag called H3K4me1 (often thought of as “primed”) carried the highest levels of both methyl and hydroxymethyl marks on DNA, especially when examined directly at H3K4me1-bound nucleosomes. In contrast, enhancers with added signs of strong activity or repression carried less of these DNA marks or showed a shift toward hydroxymethylation, hinting at ongoing erasure of methyl marks.
Decoding Enhancer States with Finer Detail
Because all enhancer types share the H3K4me1 mark, the team asked whether the detailed pattern of DNA marks specifically at H3K4me1-tagged DNA could by itself tell different enhancer states apart. They trained a machine-learning model using the 6-base-CUT&Tag data to classify enhancers as active, primed, or poised, based purely on how much methyl and hydroxymethyl they carried at that single histone feature. This model outperformed an otherwise identical model trained on standard, whole-genome data that is not restricted to any histone mark. In other words, reading DNA marks in the immediate context where they occur gives a sharper picture than averaging across all DNA in the cell.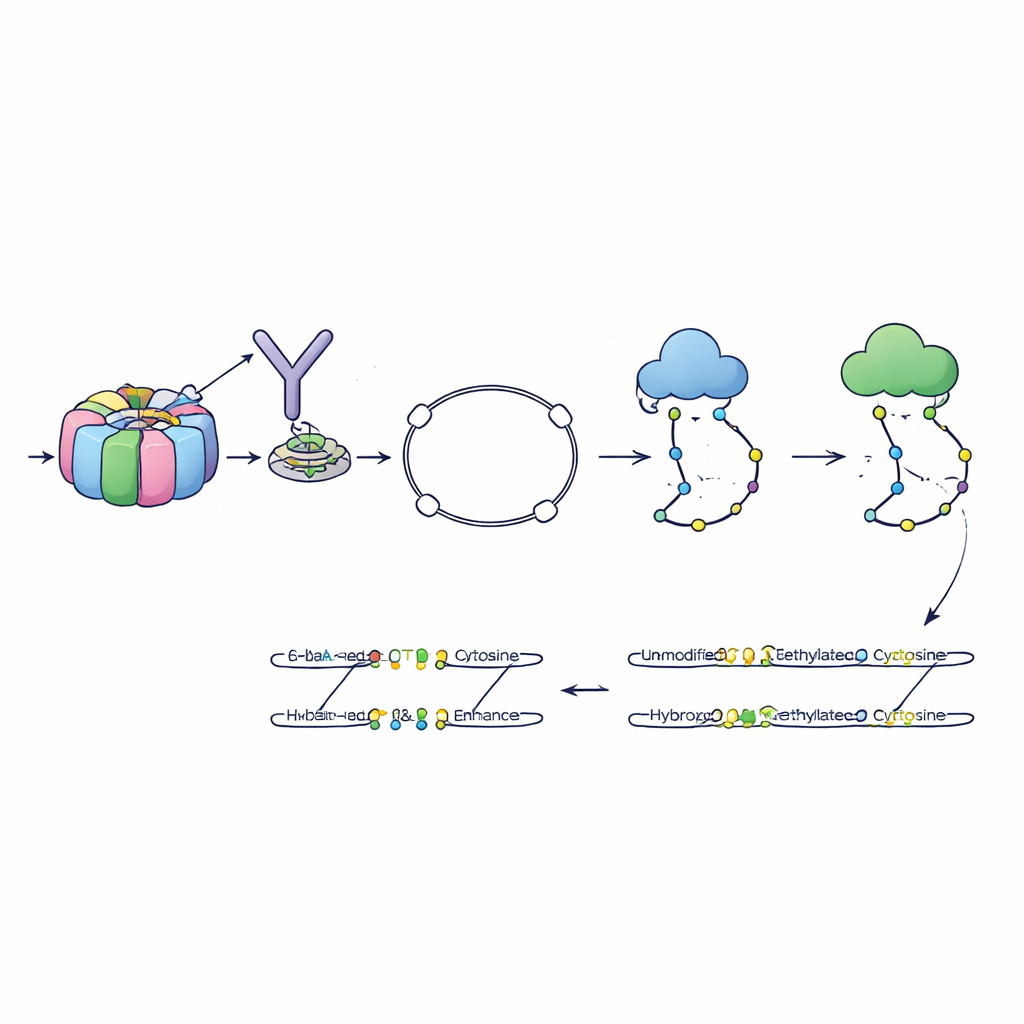
What This Means for Understanding Cell Identity
To a non-specialist, the key message is that this method lets scientists read several layers of information—DNA sequence, DNA marks, and histone marks—on the very same molecule. This fine-grained view reveals how particular combinations of chemical tags define the readiness of gene switches in stem cells. Because 6-base-CUT&Tag is more efficient and less damaging than older approaches, it can uncover subtle patterns that were previously hidden. Over time, this multi-layer reading of chromatin could help explain how cells remember their identity, how they change during development or disease, and how we might more precisely target the regulatory code in therapies.
Citation: Araujo Tavares, R.d.C., Dhir, S., He, X. et al. Sequencing DNA methylation and hydroxymethylation at co-occurring chromatin features. Nat Commun 17, 2591 (2026). https://doi.org/10.1038/s41467-026-69429-6
Keywords: epigenetics, DNA methylation, chromatin, enhancers, stem cells