Clear Sky Science · en
Structural defects in amyloid-β fibrils drive secondary nucleation
Why tiny flaws in brain proteins matter
In Alzheimer’s and related brain diseases, certain proteins clump together into long, thread-like structures called amyloid fibrils. These fibrils not only mark the disease; they also help generate new, highly toxic protein particles that can damage brain cells. This study asks a simple but powerful question: do rare structural "flaws" inside amyloid fibrils act as the main hot spots that spark new harmful growth? The answer could point to new ways to slow or stop these disorders by targeting just a few critical sites instead of an entire protein surface. 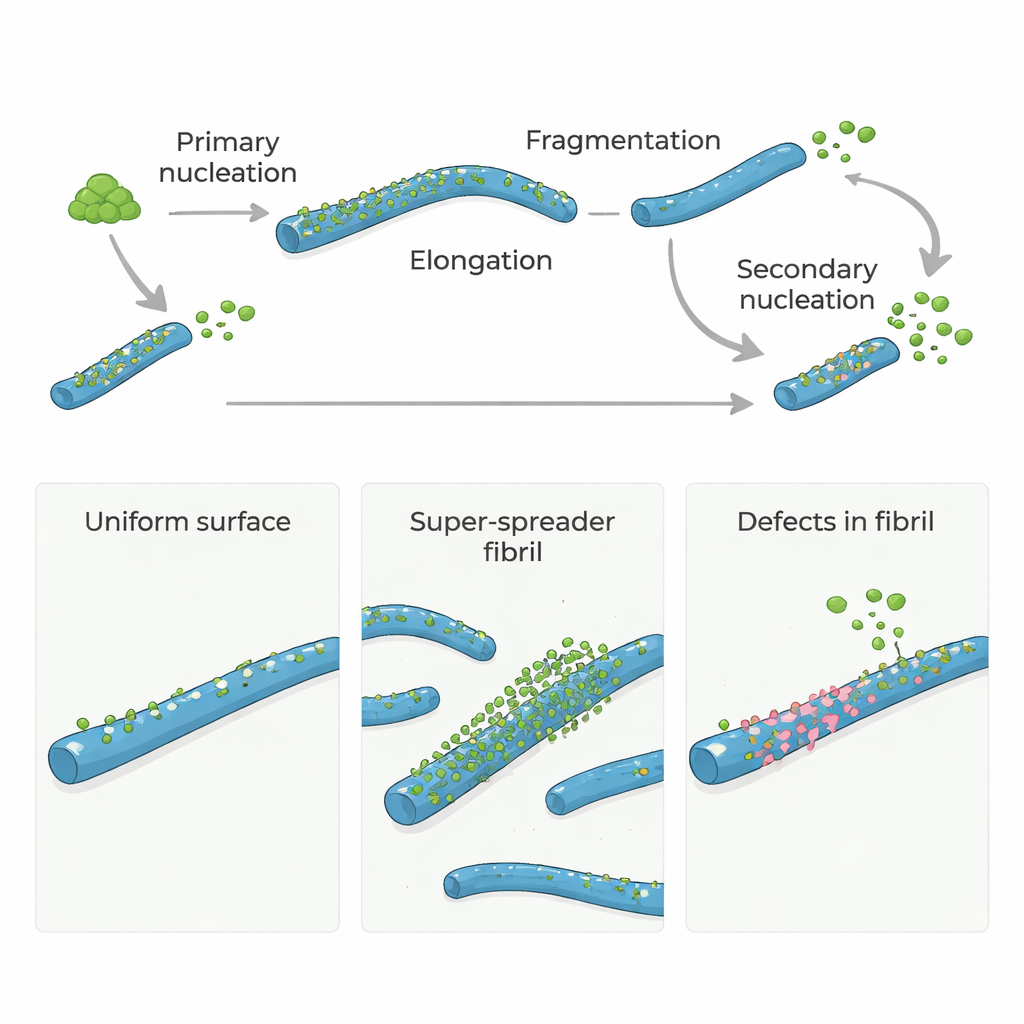
From quiet protein to runaway chain reaction
Amyloid-β (Aβ) proteins, central to Alzheimer’s disease, do not normally form clumps on their own very quickly. To get started, a few monomers must slowly assemble into the first tiny fibrils, a step called primary nucleation. Once these initial fibrils exist, they grow rapidly as more monomers add to their ends. Even more important, existing fibrils can trigger new fibrils to appear on their surfaces in a process known as secondary nucleation. This surface-driven step can flood the system with new fibrils and small, toxic oligomers, turning a slow trickle into a runaway chain reaction.
Are all fibril surfaces equally dangerous?
Many models have assumed that the entire surface of an amyloid fibril is equally good at catalyzing secondary nucleation. However, recent experiments hinted that only a small fraction of the surface is truly active. To probe this, the authors used a natural molecular "chaperone" called Brichos, known to block secondary nucleation of Aβ40 and Aβ42 (two key forms of amyloid-β). By carefully measuring how much fluorescently labeled Brichos sticks to fibrils, they found that it binds tightly but at very low numbers: roughly one Brichos molecule for every 100–150 Aβ molecules in a fibril. Yet this sparse coverage was enough to suppress more than 90% of secondary nucleation, meaning that only rare, localized sites—rather than the whole surface—dominate the production of new toxic assemblies.
Probing the role of hidden defects
These findings suggested that the crucial nucleation sites might be structural defects—small irregularities formed as fibrils grow, such as misaligned layers or partially exposed inner cores. To test this idea directly, the researchers grew Aβ40 fibrils under two different conditions. One set, the "control" fibrils, was formed under typical, strongly supersaturated conditions that favor rapid growth and kinetic trapping of defects. The other set was created using a slow, temperature-controlled annealing protocol: fibrils were grown at very low effective driving force, close to their solubility limit, where incorrect structures can dissolve or repair before becoming locked in. High-resolution cryo-electron microscopy showed that both sets of fibrils looked essentially identical in overall shape and twist, indicating that the annealing process did not change the basic morphology. 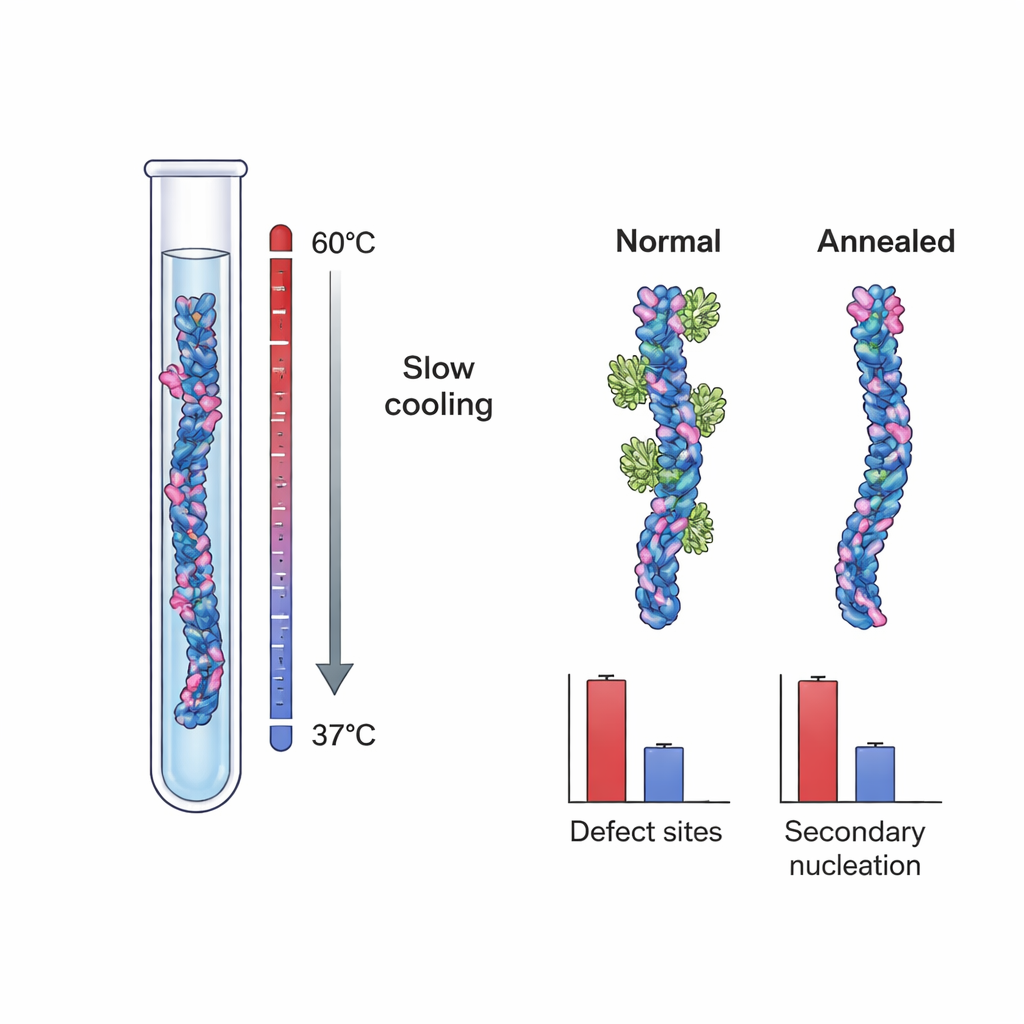
Fewer flaws, fewer dangerous growth sites
When the team measured Brichos binding to these two fibril types, they discovered a striking difference. Control Aβ40 fibrils had about one Brichos-binding site per ~100 monomers, again indicating rare but important sites. Annealed fibrils, however, had only about one site per ~800 monomers—an almost 90% reduction in site frequency. In separate tests where these fibrils were added as “seeds” to fresh Aβ40 solutions, annealed fibrils were much less effective at triggering new aggregation, even when their total mass was matched to that of control fibrils. Detailed kinetic modeling showed that this drop in seeding power could not be explained simply by differences in fibril length. Instead, it lined up quantitatively with the reduced number of Brichos-binding sites, strongly supporting the idea that growth defects act as the main engines of secondary nucleation.
A general principle with therapeutic promise
By combining thermodynamic arguments, re-analysis of earlier work, and comparisons across several amyloid-forming proteins, the authors argue that rare growth defects are likely to be central to secondary nucleation in many systems, not just Alzheimer’s-related Aβ. These defects partially expose the tightly packed inner core of the fibril, offering a ready-made scaffold where new oligomers and fibrils can form far more easily than on a smooth surface. Recognizing these defects as the key culprits opens new avenues for drug design. Instead of trying to block every possible interaction on a fibril surface, therapies could aim to shield or repair just these scarce defect sites, or to reduce the conditions that create them in the first place. In practical terms, that might mean lowering the effective concentration of amyloid-forming proteins in the brain or designing molecules, inspired by Brichos, that recognize and neutralize defect-driven nucleation hot spots. If successful, such strategies could cut off the main source of toxic oligomers and slow the progression of amyloid-linked diseases.
Citation: Hu, J., Scheidt, T., Thacker, D. et al. Structural defects in amyloid-β fibrils drive secondary nucleation. Nat Commun 17, 1933 (2026). https://doi.org/10.1038/s41467-026-69377-1
Keywords: amyloid fibrils, Alzheimer’s disease, secondary nucleation, protein aggregation, Brichos chaperone