Clear Sky Science · en
STING synergizes with TOX suppressing HO-1 expression to trigger ferroptosis in tumor-infiltrating CD8+ T cell and immunotherapy resistance
Why this research matters for cancer treatment
Modern cancer immunotherapies work by unleashing the body’s own killer T cells, yet many tumors still find ways to shut these cells down. This study uncovers a hidden self-destruct switch inside CD8+ “killer” T cells that tumors exploit, and shows how turning that switch off can make immunotherapy far more powerful.
A hidden death pathway inside tumor-fighting T cells
Inside a tumor, CD8+ T cells are supposed to hunt and destroy cancer cells. Instead, they often become scarce, sluggish, and short-lived. The authors focused on two molecules inside T cells—STING, a sensor of DNA damage, and TOX, a protein linked to T cell exhaustion. They engineered mice in which CD8+ T cells lacked STING, TOX, or both, and then implanted several types of tumors. Surprisingly, mice whose T cells were missing either STING or TOX cleared tumors much better. Their tumors grew more slowly, contained many more CD8+ T cells, and these T cells produced larger amounts of cancer-killing molecules such as interferon-gamma and granzyme B. This pointed to an internal program that was quietly sabotaging T cells inside tumors.
How iron-driven cell death weakens immunity
By examining gene activity in tumor-infiltrating T cells, the researchers found that normal T cells inside tumors were primed for a particular form of cell death called ferroptosis. Unlike apoptosis, ferroptosis is triggered by iron overload and the buildup of damaged fats in cell membranes. In ordinary CD8+ T cells exposed to tumor cells, genes that promote iron accumulation and lipid damage were switched on, while protective genes were turned down. In contrast, STING- or TOX-deficient T cells showed the opposite pattern: they expressed more of the protective enzymes HO-1 and GPX4, had lower iron levels, less lipid peroxidation, healthier mitochondria, and resisted ferroptotic death. Lab tests confirmed that blocking ferroptosis chemically kept normal T cells alive, whereas removing STING or TOX made them naturally resistant.
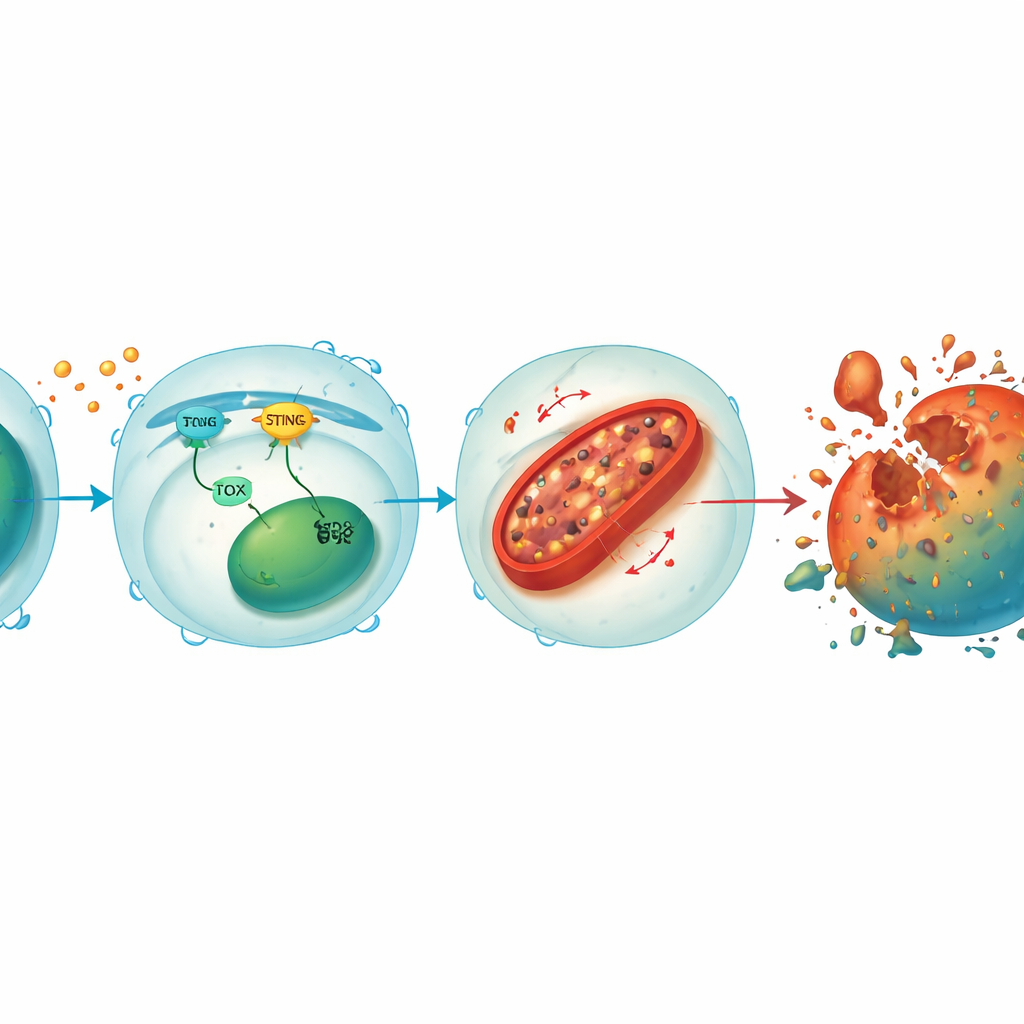
A loop that links stress signals to mitochondrial damage
Diving deeper, the team discovered that STING and TOX form a reinforcing loop inside CD8+ T cells. When tumor or viral signals activate STING, it triggers downstream factors that boost TOX, and TOX in turn helps maintain STING activity. Together, they suppress HO-1, an enzyme that normally helps keep cellular iron in check. With HO-1 held down, iron builds up in mitochondria—the cell’s power plants—leading to high levels of reactive oxygen molecules and oxidation of membrane fats. This mitochondrial damage drains energy production and ultimately pushes the T cell into ferroptosis. Restoring STING or TOX in knockout T cells brought back iron overload and cell death, while further lowering HO-1 made even the protected T cells vulnerable again, underscoring HO-1 as a central brake on this destructive pathway.
How tumor-made lactate pulls the trigger
The tumor microenvironment is rich in lactate, a byproduct of cancer’s altered metabolism. The study shows that this lactate is not just metabolic waste—it actively helps flip the ferroptosis switch in T cells. Compared with other cells, CD8+ T cells were especially sensitive to lactate. As lactate entered the cells through specific transporters, it caused iron buildup, mitochondrial shrinkage, DNA loss in mitochondria, and more oxidative damage. At the same time, lactate boosted STING and TOX activity and further suppressed HO-1. T cells lacking STING or TOX were far more resistant to lactate-induced injury. Blocking a key lactate transporter with a drug (AZD3965) protected CD8+ T cells from ferroptosis in mice, increased their presence inside tumors, and slowed tumor growth, mimicking the benefit of genetically deleting STING in T cells.
Turning a vulnerability into a therapeutic advantage
These mechanistic insights have practical implications. When the researchers used adoptive cell therapy—infusing mice with lab-activated T cells—they found that T cells engineered to lack STING or TOX produced much stronger tumor control than normal T cells. Moreover, combining these “ferroptosis-resistant” T cells with existing treatments such as PD-1 or TIM-3 checkpoint blockers, cisplatin chemotherapy, or a STING-activating drug yielded markedly better tumor shrinkage than any single therapy alone. Finally, in tumor samples from patients with cervical cancer, higher TOX and lower HO-1 levels in tumor-infiltrating lymphocytes were linked to poorer survival, suggesting that this pathway also shapes clinical outcomes in people.
What this means for future cancer care
In plain terms, the study reveals that tumors can force our best cancer-fighting T cells to rust from the inside out by driving an iron-fueled form of cell death. A lactate–STING–TOX circuit lowers the protection offered by HO-1, damages mitochondria, and leads to ferroptosis, thinning the ranks of effective CD8+ T cells. Disrupting this circuit—by engineering T cells that lack STING or TOX, boosting HO-1, or blocking lactate entry—keeps T cells alive, energetic, and ready to attack. This work points toward next-generation immunotherapies that combine metabolic and genetic tuning of T cells with existing drugs to overcome resistance and deliver more durable cancer control.
Citation: Zhu, Q., Zhang, Jb., Nie, Cp. et al. STING synergizes with TOX suppressing HO-1 expression to trigger ferroptosis in tumor-infiltrating CD8+ T cell and immunotherapy resistance. Nat Commun 17, 2543 (2026). https://doi.org/10.1038/s41467-026-69350-y
Keywords: cancer immunotherapy, CD8 T cells, ferroptosis, tumor microenvironment, STING TOX HO-1 pathway