Clear Sky Science · en
CD38 degrades MAVS through mitophagy to inhibit type I interferon secretion in nasopharyngeal carcinoma cells and impairs CD8+T cell-mediated anti-tumor immunity
Why this matters for cancer treatment
Nasopharyngeal carcinoma is a cancer that arises behind the nose and is especially common in East and Southeast Asia. Immune-based drugs that unleash the body’s own T cells have changed the outlook for some patients, but most still do not benefit. This study uncovers a hidden brake inside tumor cells themselves: a molecule called CD38 that quietly shuts down an internal alarm system and weakens the attack of cancer-killing CD8 T cells. Understanding and disabling this brake could make existing immunotherapies work for many more people.
A hidden switch on tumor cells
The researchers focused on CD38, a protein found on many immune cells but also present on nasopharyngeal carcinoma cells. Earlier work had linked CD38 to resistance against popular checkpoint drugs that target PD-1 and PD-L1. Here, the team asked whether CD38 inside tumor cells directly alters how well CD8 T cells can recognize and destroy those cells. By growing human tumor cells with or without CD38 alongside activated human CD8 T cells, they found that removing CD38 from the cancer cells made the T cells far more potent: they secreted higher levels of key attack molecules, survived better, and killed more tumor cells. When CD38 was added back, T cell function dropped, pointing to CD38 as a tumor-intrinsic suppressor of immune attack.
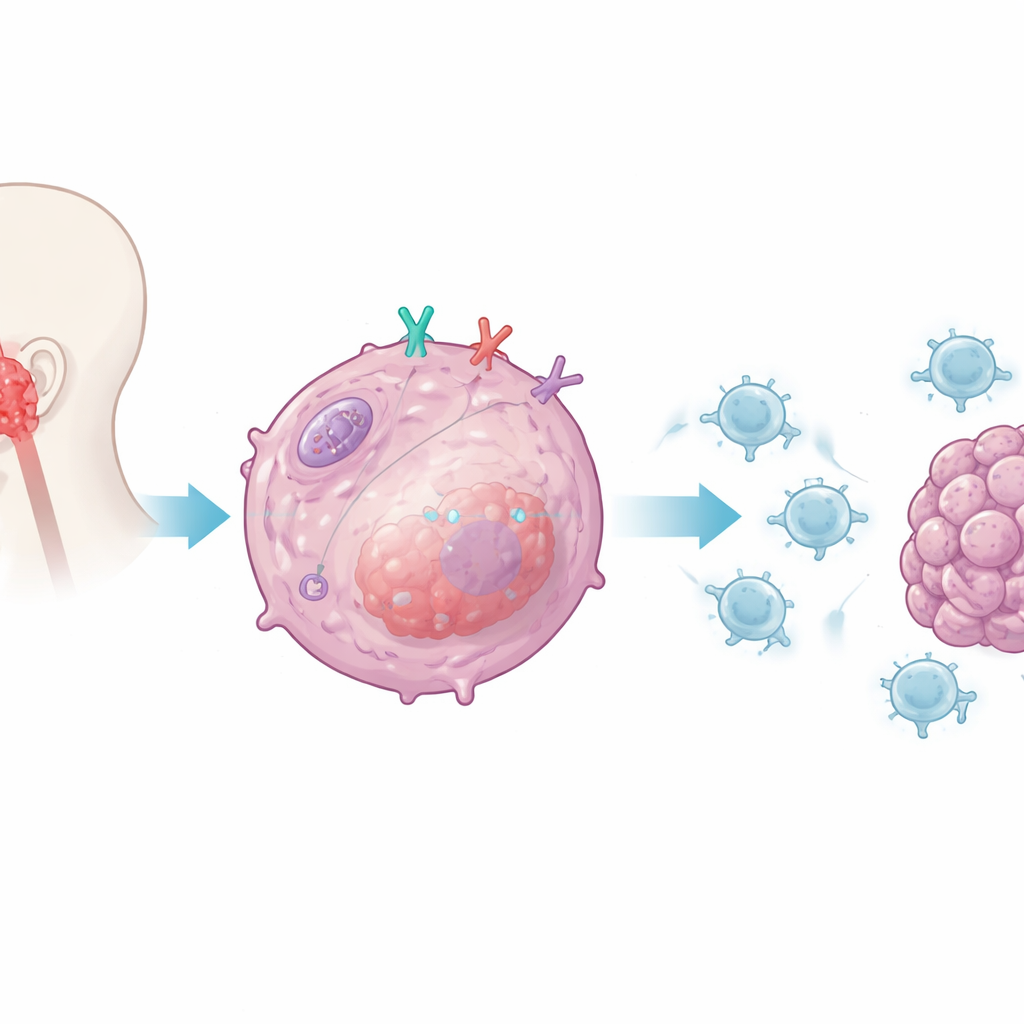
Turning down the cell’s internal alarm
The team then investigated how CD38 sends this suppressive signal. They focused on the tumor cell’s innate alarm system, which normally detects virus-like genetic material and triggers type I interferons—potent immune-stimulating messengers. In tumor cells lacking CD38, the researchers observed a strong rise in interferon-beta and in chemokines that draw CD8 T cells into tumors. They showed that CD38 selectively dampens the pathway controlled by an internal sensor named RIG-I and its adaptor protein MAVS, which sits on mitochondria, the cell’s energy factories. When CD38 was present, activation of this pathway and its downstream signaling molecules was blunted; when CD38 was removed, signalling and interferon production surged, boosting the tumor’s visibility to the immune system.
How CD38 destroys a key signaling hub
Digging deeper, the scientists found that CD38 physically associates with MAVS on mitochondria and interferes with MAVS’s partnership with RIG-I, weakening signal transmission. More strikingly, higher CD38 levels led to a reduction in MAVS protein without changing its genetic blueprint, suggesting active destruction. Tests using different inhibitors showed that this loss depended on the cell’s recycling machinery known as autophagy, and specifically on a form that targets mitochondria. CD38 increased markers of mitochondrial “self-eating,” reduced several mitochondrial proteins, and promoted the packaging of MAVS into autophagosome structures that are later broken down. Blocking mitochondrial autophagy preserved MAVS and restored interferon signaling, indicating that CD38 disables the alarm by driving MAVS into the cell’s waste stream.
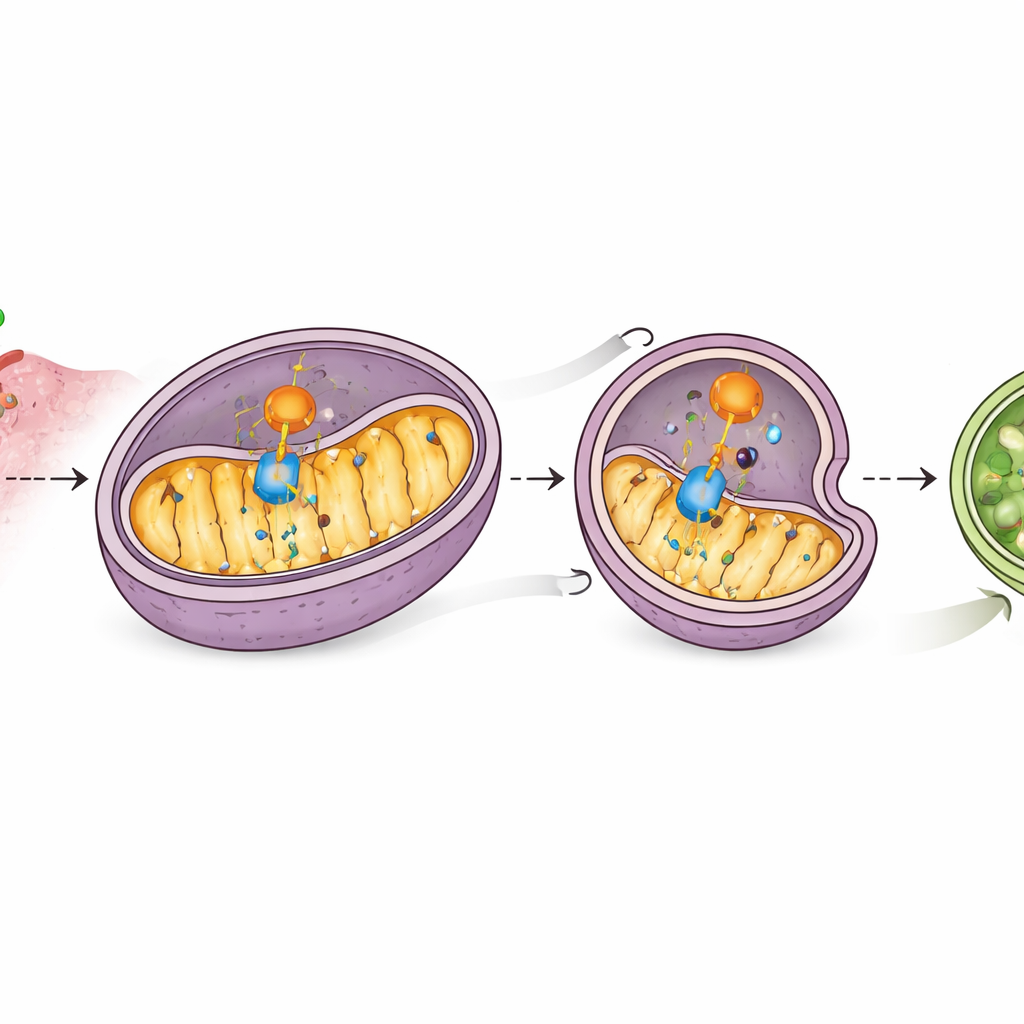
A helper that guides mitochondria to self‑destruct
The study identified another player, PHB2, a protein inside mitochondria that acts as a receptor for targeted mitochondrial removal. Mass spectrometry and binding experiments showed that CD38 interacts with PHB2 and increases PHB2’s presence in mitochondria, where PHB2 in turn recruits the core autophagy machinery. PHB2 also binds MAVS, and CD38 strengthens this contact. When PHB2 was reduced, CD38 could no longer trigger mitochondrial autophagy effectively, MAVS levels rebounded, and interferon-related genes were reactivated. This reveals a chain of events: CD38 engages PHB2, PHB2 engages MAVS, and together they usher MAVS into mitochondria destined for degradation, silencing the interferon alarm.
Evidence from animal models
To test the impact in living organisms, the researchers used mouse tumors engineered to lack CD38. In immune-competent mice, these tumors grew more slowly, contained more CD8 T cells, and had a higher fraction of cells producing interferon-gamma, a hallmark of active anti-tumor responses. Blocking the receptor for type I interferons removed this advantage, confirming that interferon signaling is essential for the enhanced immunity. In humanized mice bearing nasopharyngeal tumors, reducing CD38 similarly slowed growth and increased CD8 T cell infiltration, but this benefit vanished when MAVS was also reduced in the tumor cells. These in vivo findings cement the idea that the CD38–PHB2–MAVS axis inside tumor cells shapes the strength of the body’s T cell response.
What this means for future therapies
Overall, the work shows that CD38 in nasopharyngeal carcinoma cells acts as an internal saboteur of anti-tumor immunity. By driving a selective form of mitochondrial recycling, CD38 depletes MAVS, weakens type I interferon production, lowers antigen presentation, and ultimately blunts CD8 T cell attack. Current CD38-blocking compounds mainly target its enzyme activity and do not remove the protein or restore MAVS. The authors argue that new strategies aimed at reducing CD38 levels or disrupting its partnership with PHB2 or MAVS could reawaken the interferon alarm inside tumors. Combined with existing checkpoint inhibitors, such approaches might turn more nasopharyngeal—and potentially other—cancers from immune-cold into immune-responsive.
Citation: Liang, L., Li, W., Liu, S. et al. CD38 degrades MAVS through mitophagy to inhibit type I interferon secretion in nasopharyngeal carcinoma cells and impairs CD8+T cell-mediated anti-tumor immunity. Nat Commun 17, 2544 (2026). https://doi.org/10.1038/s41467-026-69339-7
Keywords: nasopharyngeal carcinoma, tumor immunotherapy, type I interferon, CD8 T cells, mitophagy