Clear Sky Science · en
Mycobacterium tuberculosis modulates phosphorylation of host ATP6V1E1 to promote intracellular survival
Why this matters for fighting tuberculosis
Tuberculosis remains one of the world’s deadliest infectious diseases, killing more than a million people each year. Our immune cells are equipped with powerful “acidic recycling bins” that normally digest invading microbes. This paper uncovers how the tuberculosis bacterium, Mycobacterium tuberculosis (Mtb), sabotages that acidification system inside our cells, and shows that a drug targeting this trick can help infected animals clear the infection more effectively.
The cell’s acid bath for germs
When tuberculosis bacteria are inhaled into the lungs, they are quickly swallowed by immune cells called macrophages. The bacteria end up in membrane bubbles that are supposed to fuse with lysosomes—small sacs filled with digestive enzymes that work best in a strongly acidic environment. That acidity is created by a molecular pump, the vacuolar ATPase (V-ATPase), which uses cellular fuel to push protons into lysosomes and drive the internal pH down. Proper acidification is crucial for breaking down Mtb, yet decades of work have shown that this pathogen somehow keeps its compartments less acidic and survives.
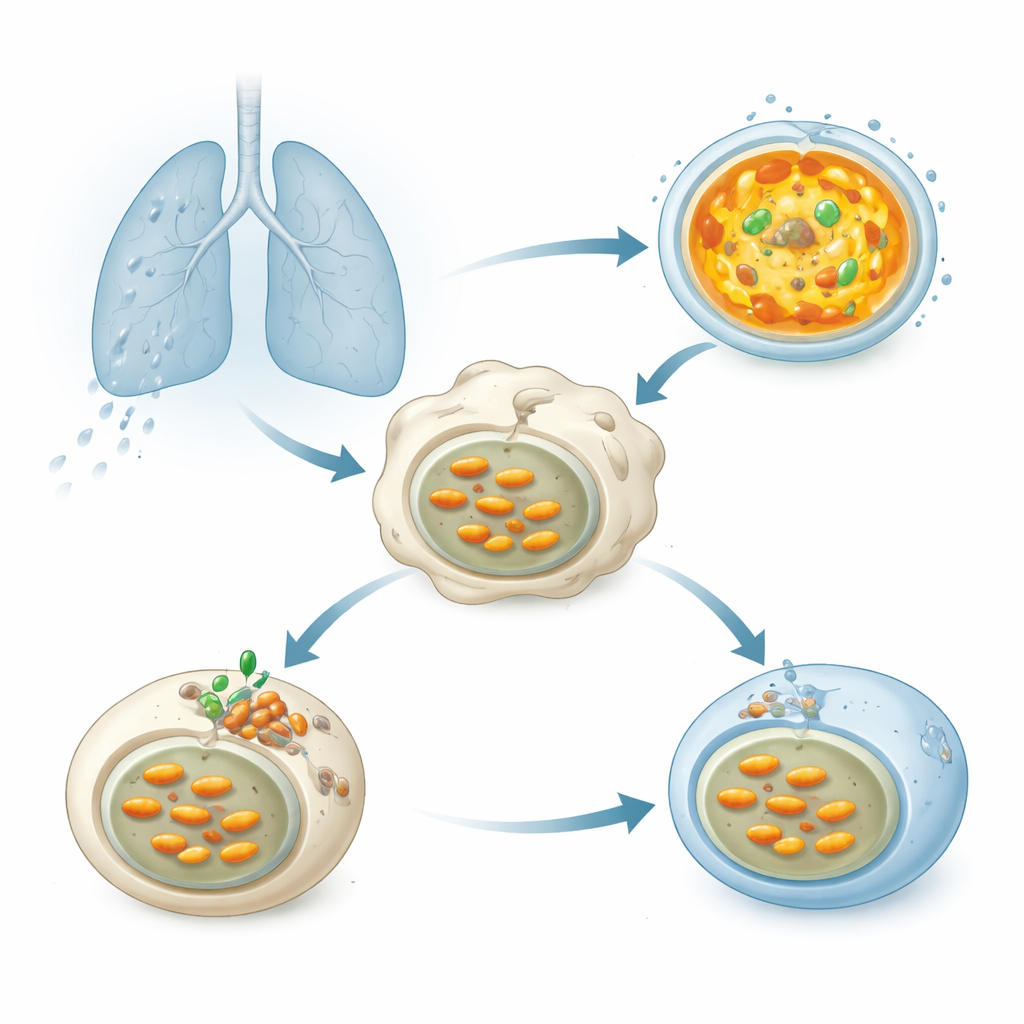
A secret bacterial helper that blocks acidification
The researchers screened more than 200 proteins that Mtb can release to see which ones weaken lysosomal acidification in human cells. One stood out: an enzyme called Chp2 (also known as Rv1184). When cells produced Chp2, their lysosomes lit up more weakly with acid-sensitive dyes, indicating a higher, less hostile pH. Mtb strains engineered to lack Chp2 no longer suppressed acidification; in infected macrophages and in mice, these mutant bacteria were cleared more efficiently and caused milder lung damage. Restoring Chp2 to the mutant strain brought back both the acidification block and the higher bacterial burden, identifying Chp2 as a virulence factor that helps Mtb persist inside host cells.
The host pump subunit that acts as a control switch
To understand how Chp2 interferes with acidification, the team looked for host components it might bind. They found that Chp2 directly attaches to a particular part of the V-ATPase, a subunit called ATP6V1E1 (E1 for short), which helps stabilize the pump’s structure. Increasing the amount of E1 in cells boosted lysosomal acidification and made it harder for Mtb to survive, whereas reducing E1 had the opposite effect. Mice that carried only one working copy of the E1 gene had less acidic lysosomes, higher bacterial loads in their lungs, and more severe tissue damage after infection, showing that E1 is an important host defender against tuberculosis.
A kinase-tag on the pump that turns down the acid
The authors then asked whether chemical “tags” on E1 might tune the pump’s activity. They discovered that adding phosphate groups to two specific tyrosines (Tyr56 and Tyr57) on E1 acts like a brake: mimicking phosphorylation reduced acidification and prevented complete assembly of the V-ATPase, while preventing phosphorylation had the opposite effect. By screening a panel of enzymes, they identified a kinase called BMX as the host protein that places this tag. When BMX was blocked genetically or with a small-molecule inhibitor, E1 phosphorylation dropped, the pump assembled more efficiently on lysosomal membranes, lysosomes became more acidic, and Mtb survival inside macrophages decreased.
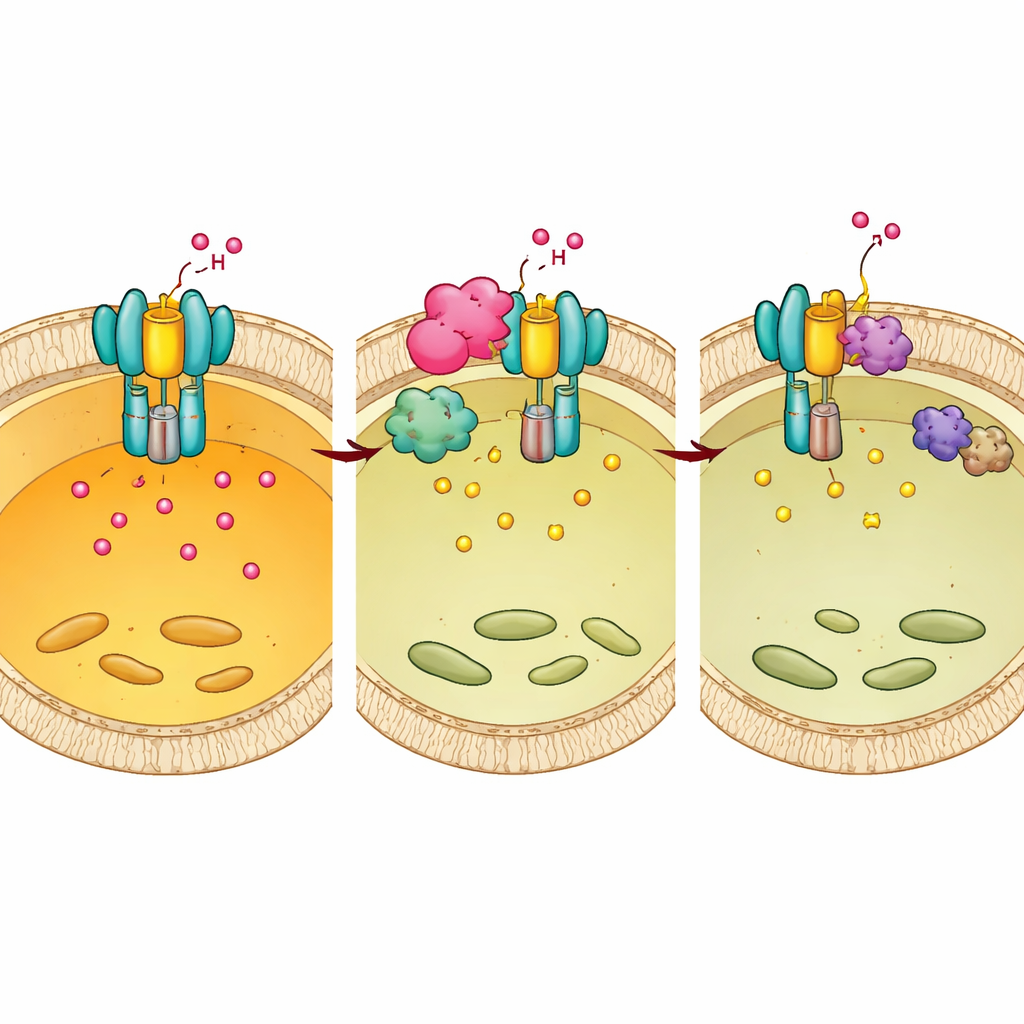
How Mtb rewires the host switch in its favor
Chp2 and BMX turn out to work together. Structural and biochemical experiments showed that Chp2 acts as a scaffold that brings E1 and BMX into close contact on the lysosomal surface, enhancing E1 phosphorylation at Tyr56/57. This extra tagging disrupts full pump assembly, raises lysosomal pH, and creates a gentler environment in which Mtb can persist. When BMX was inhibited, the survival advantage conferred by Chp2 disappeared, both in cultured cells and in infected mice. Importantly, treating mice with the BMX inhibitor after infection reduced bacterial loads and lung pathology in normal animals, but not in mice with weakened E1, indicating that the drug works by restoring effective E1-driven acidification.
Turning the pathogen’s trick into a treatment idea
In everyday terms, this study shows that tuberculosis bacteria slip a helper protein into our cells that meddles with the “acid pump” on the garbage disposal, turning it down just enough for the microbes to ride out destruction. By identifying the pump subunit that serves as the key control knob (E1) and the host enzyme that flips it (BMX), the authors reveal a precise point where a drug can intervene. Inhibiting BMX in mice effectively reactivates the cell’s internal acid bath and improves bacterial clearance. These findings open the door to host-directed therapies that make our own cells more hostile to Mtb, potentially working alongside antibiotics and helping to combat drug-resistant tuberculosis.
Citation: Chen, J., Tang, F., Qin, L. et al. Mycobacterium tuberculosis modulates phosphorylation of host ATP6V1E1 to promote intracellular survival. Nat Commun 17, 2434 (2026). https://doi.org/10.1038/s41467-026-69331-1
Keywords: tuberculosis, lysosomes, host-directed therapy, Mycobacterium tuberculosis, V-ATPase