Clear Sky Science · en
Histone lactylation increases CXCL1 expression for neutrophil infiltration and immune escape in pancreatic cancer
Why this cancer study matters
Pancreatic cancer is one of the deadliest cancers, in part because it often resists modern immunotherapies that work in other tumors. This study uncovers how pancreatic tumors rewire their own sugar metabolism to quietly disarm the immune system, and how blocking that process may reopen a path for the body’s defenses—and existing drugs—to fight back.
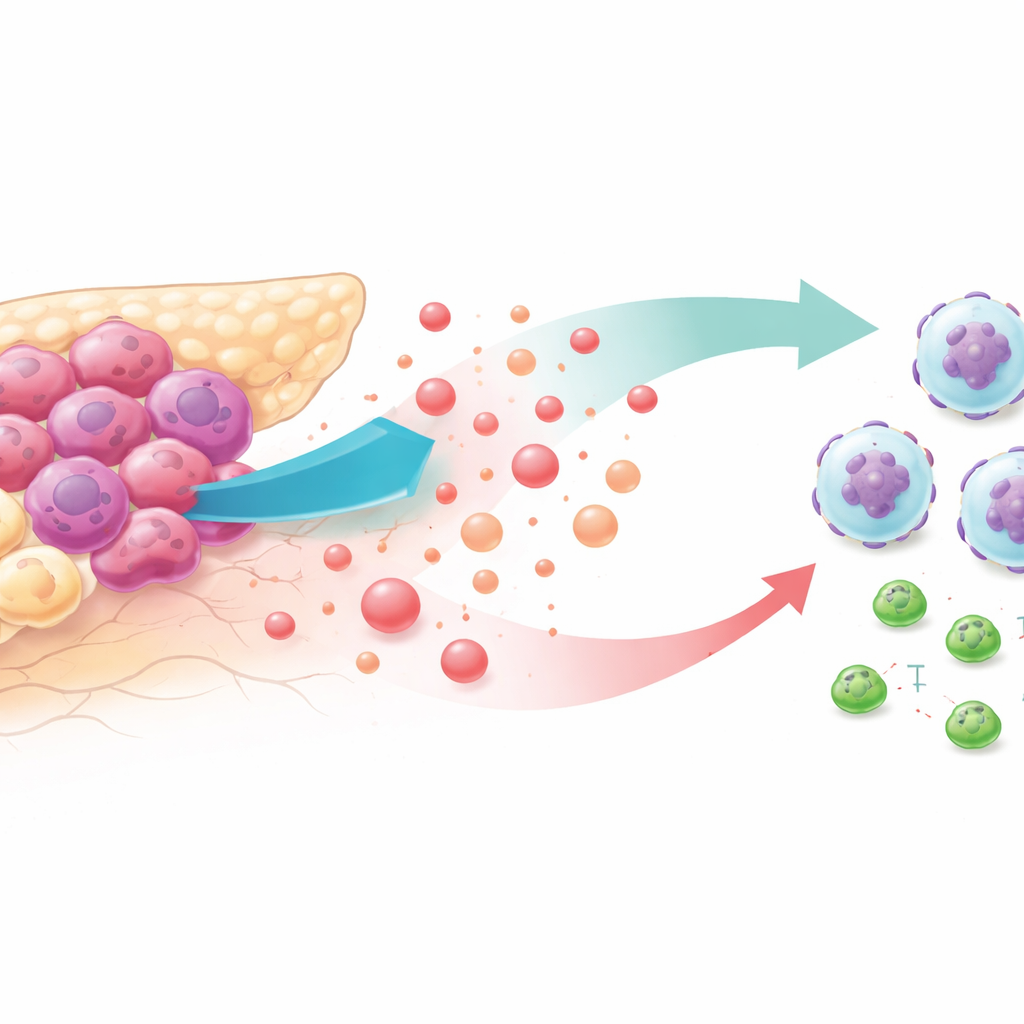
Sugar-hungry tumors and a hostile neighborhood
Cancer cells are notorious for burning glucose at a furious pace, even when oxygen is plentiful. This "high-glycolysis" lifestyle floods the tumor neighborhood with lactate, a byproduct once dismissed as metabolic waste. By analyzing patient tumor databases and mouse models, the researchers found that pancreatic cancers with the most intense glycolysis were packed with neutrophils—white blood cells that, in this context, help tumors grow—and had fewer cancer-killing CD8 T cells. Patients whose tumors showed this profile tended to live for a shorter time, suggesting that altered metabolism and immune escape are tightly linked.
How tumors use lactate to call in the wrong help
To understand how glycolysis attracts neutrophils, the team turned down tumor glycolysis with drugs or genetic tricks in pancreatic cancer cell lines and mice. When sugar breakdown was blocked, tumor cells released much less of a chemical signal called CXCL1, and circulating levels of this signal dropped in mice and in patient samples. In laboratory migration tests, neutrophils moved eagerly toward media from highly glycolytic cancer cells but not toward media from glycolysis-blocked cells—unless the researchers added back purified CXCL1. In living mice, restoring CXCL1 in tumors that had reduced glycolysis brought neutrophil numbers back up and weakened the anti-tumor effect of glycolysis inhibition.
A new epigenetic switch driven by lactate
The study then zoomed in to the level of DNA packaging. Our genes are wrapped around spool-like proteins called histones, whose chemical tags act as on/off switches for gene activity. The authors show that in pancreatic cancer, lactate produced by glycolysis adds a specific "lactyl" tag to one histone position, known as H3K18. This modification, called histone H3K18 lactylation, was markedly higher in tumor tissues than in normal pancreas. When glycolysis was blocked, lactylation at H3K18 fell, especially near the CXCL1 gene’s control region, and CXCL1 production dropped. Adding back lactate restored both the histone tag and CXCL1 expression. Across patient samples, tumors with more H3K18 lactylation also showed higher CXCL1, linking this molecular mark to a pro–tumor immune landscape.
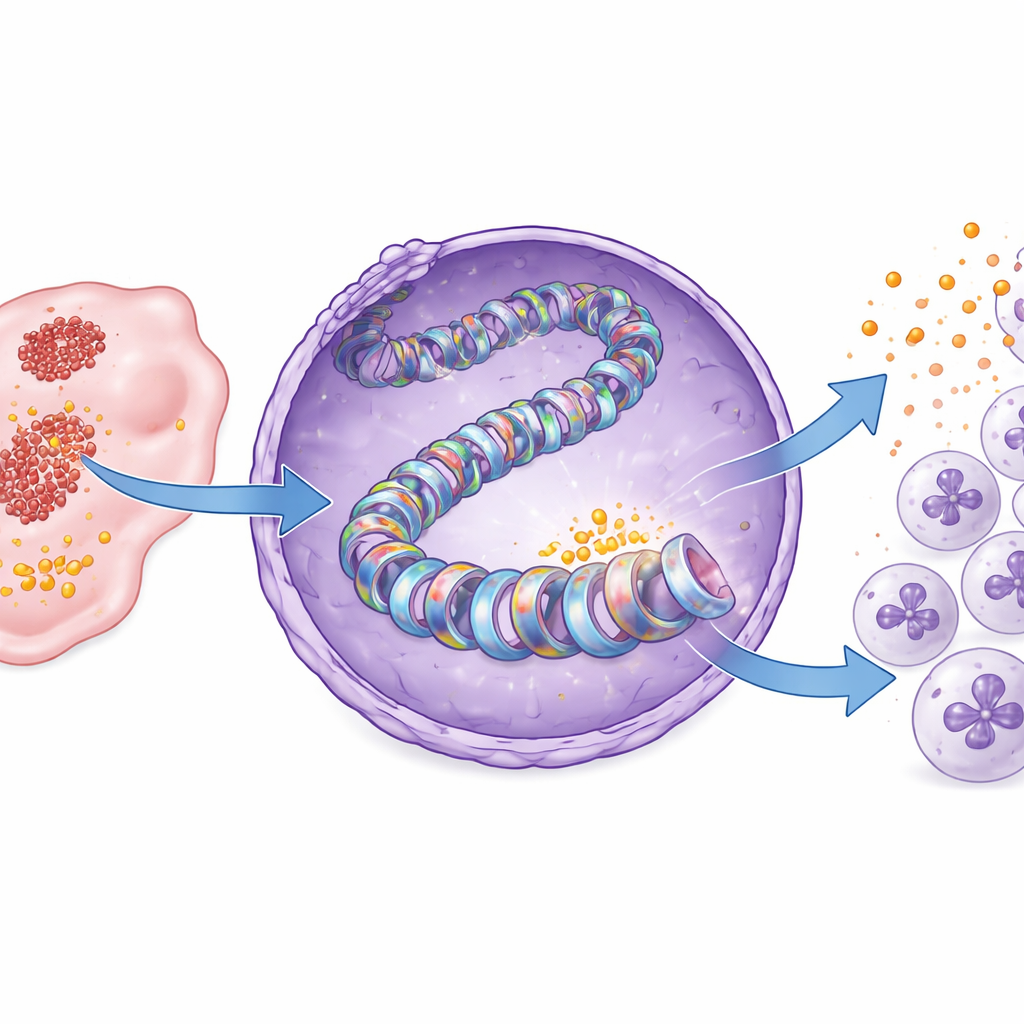
Identifying the enzyme and a druggable weak spot
Histone tags are written by specialized enzymes. By screening inhibitors of known histone-modifying proteins, the researchers pinpointed one called PCAF as a key writer of the H3K18 lactylation mark in pancreatic cancer. Structural modeling suggested that PCAF can bind lactyl-CoA, the activated form of lactate used for tagging, and biochemical assays confirmed that purified PCAF can directly add lactyl groups to histone H3. Blocking PCAF with a small molecule, bromosporine, lowered H3K18 lactylation and CXCL1 production in cancer cells and in mouse tumors. As a result, fewer neutrophils entered the tumors, more CD8 T cells accumulated, and tumor growth slowed, all without obvious weight loss or toxicity in the mice.
Turning a cold tumor hot with combination therapy
Because standard immune checkpoint drugs such as anti–PD-1 antibodies have shown limited success in pancreatic cancer, the team tested whether dismantling the lactate–PCAF–CXCL1 pathway could make these tumors more responsive. In both subcutaneous and pancreatic orthotopic mouse models, bromosporine combined with anti–PD-1 therapy shrank tumors more than either treatment alone, reduced neutrophil infiltration, boosted active CD8 T cells, and significantly prolonged survival. This suggests that cutting the tumor’s metabolic "siren" for neutrophils helps turn an immunologically "cold" tumor into a "hotter" one that checkpoint therapy can better engage.
What this means for future treatment
In plain terms, the study reveals a chain reaction: pancreatic tumors burn sugar, release lactate, use that lactate to flip an epigenetic switch on their DNA-packaging proteins, crank up CXCL1, and thereby summon neutrophils that help them hide from killer T cells. Interrupting this chain at the PCAF step with a drug not only slows tumors but also makes existing immunotherapy more potent in mice. While more work is needed to ensure safety and find the best way to target this pathway in people, these findings highlight a promising strategy: reprogram the tumor’s metabolism and gene switches to give the immune system a fair fight.
Citation: Zhang, P., Ma, J., Wan, Y. et al. Histone lactylation increases CXCL1 expression for neutrophil infiltration and immune escape in pancreatic cancer. Nat Commun 17, 2526 (2026). https://doi.org/10.1038/s41467-026-69311-5
Keywords: pancreatic cancer, tumor metabolism, histone lactylation, tumor microenvironment, cancer immunotherapy