Clear Sky Science · en
Targeting NHEJ activates STING signaling through MYC degradation to boost antitumor immunity in SCLC
Why this research matters
Small-cell lung cancer is one of the deadliest cancers, with most patients living less than a year after diagnosis. Strikingly, these tumors carry many DNA mutations that should make them easy targets for the immune system, yet in practice they respond poorly to modern immunotherapy drugs. This study uncovers a hidden molecular brake that keeps the immune system from recognizing these tumors and shows how turning off a key DNA repair protein can flip these cancers from immune “cold” to “hot,” allowing existing treatments to work far better.
A hidden repair switch in lung tumors
The researchers began by sifting through genetic data from more than 179,000 human tumors across 24 cancer types. They focused on a DNA repair pathway called nonhomologous end joining, which fixes dangerous breaks in DNA strands. A central controller of this pathway, a protein called DNAPKcs (encoded by the PRKDC gene), turned out to be unusually high in small-cell lung cancer. Within thousands of lung tumor samples, small-cell cases showed the strongest activity of this repair switch. Patients whose tumors had the highest PRKDC levels lived for a shorter time and were less likely to benefit from standard chemotherapy and immune checkpoint drugs, suggesting that DNAPKcs helps tumors survive both DNA damage and immune attack.
From DNA damage to an internal alarm
To see what happens when this repair switch is turned off, the team used both drugs and gene-silencing tools to block DNAPKcs in panels of small-cell lung cancer cells and in mouse tumor models. In many of these models, especially those resembling human subtypes with high MYC oncogene activity, DNAPKcs inhibitors sharply reduced tumor cell growth and even shrank patient-derived tumors in mice. At the cellular level, blocking DNAPKcs led to an accumulation of broken DNA, visible as puncta of a damage marker inside the nucleus and as tiny extra DNA-filled bodies called micronuclei. These fragments of DNA spilled into the cell’s cytoplasm, where they could be sensed as danger signals.
Turning on the cell’s “viral” alarm system
Loose DNA in the wrong place is normally a sign of viral infection. Cells detect it using a sensor called cGAS, which triggers a downstream alarm pathway named STING. The authors showed that after DNAPKcs inhibition, cGAS clustered on micronuclei, STING became activated, and a cascade of immune-stimulating molecules was turned on. The cells produced more type I and type II interferons and chemokines that attract immune cells. Surface display of key “flag” proteins (MHC class I molecules), which help immune cells recognize tumor antigens, also increased. When the STING pathway was chemically blocked or genetically silenced, these changes largely disappeared, and the anti-tumor effects of DNAPKcs inhibition were much weaker, underscoring that this internal alarm system is essential for the response.
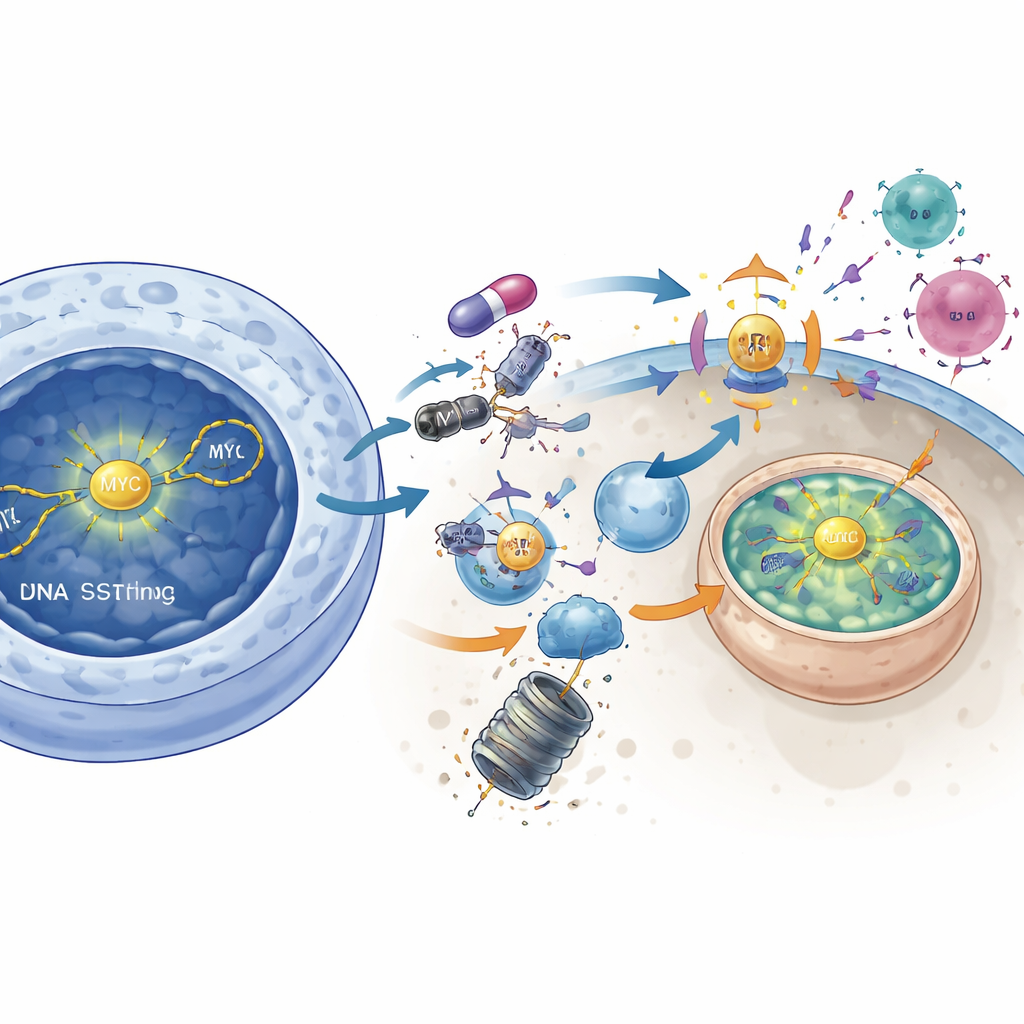
Disarming MYC to unmask the tumor
The study further links DNAPKcs to the powerful growth driver MYC, a protein long considered “undruggable.” In tumors with high MYC activity, DNAPKcs inhibition reduced active AKT signaling and released a molecular brake on another enzyme, GSK3β. Once activated, GSK3β tagged MYC for destruction, causing MYC protein levels to fall. Directly lowering MYC using genetic tools mimicked many of the immune-activating effects of DNAPKcs blockade: STING signaling rose, interferon genes turned on, and MHC class I increased. Conversely, forcing cells to overproduce MYC largely erased the immune-boosting impact of the DNAPKcs inhibitor. This suggests that DNAPKcs normally helps stabilize MYC, and that pushing MYC into degradation is a key step in waking up anti-tumor immunity.
From “cold” to “hot” tumors in living models
In immunocompetent mouse models that closely resemble human small-cell lung cancer, treatment with a DNAPKcs inhibitor alone significantly slowed or shrank tumors. Importantly, combining the inhibitor with an existing anti–PD-L1 checkpoint drug transformed previously resistant tumors, leading to dramatic tumor regression and, in some cases, complete disappearance. Detailed immune profiling showed that DNAPKcs inhibition increased cancer-killing CD8 T cells, boosted pro-inflammatory M1 macrophages, reduced suppressive T cells, and raised MHC class I levels in tumors. Removing CD8 T cells or disabling STING reversed these benefits, confirming that the therapy works by turning the tumor into a beacon for immune attack rather than by killing cancer cells directly alone.
What this means for patients
Together, these results reveal DNAPKcs as a central coordinator of both DNA repair and immune evasion in small-cell lung cancer. By blocking DNAPKcs, tumors accumulate DNA damage, MYC is destabilized, the cGAS–STING alarm is triggered, and interferon and antigen-presenting pathways are switched on. This chain of events converts immune-silent tumors into ones that respond strongly to checkpoint blockade and chemotherapy in preclinical models. While clinical trials are still needed, the work suggests that existing DNAPKcs inhibitors could be combined with immunotherapy to give patients with this aggressive cancer a better chance at long-lasting control.
Citation: Chakraborty, S., Elliott, A., Sen, U. et al. Targeting NHEJ activates STING signaling through MYC degradation to boost antitumor immunity in SCLC. Nat Commun 17, 2597 (2026). https://doi.org/10.1038/s41467-026-69262-x
Keywords: small-cell lung cancer, DNA repair inhibition, STING pathway, MYC degradation, tumor immunotherapy