Clear Sky Science · en
Pan-RAF inhibitor exarafenib targets BRAF class II/III NSCLC and reveals ARAF-KSR1 resistance and combination strategies
Why this research matters for lung cancer patients
Most people have heard of targeted cancer drugs that home in on specific genetic changes. But for many patients with lung cancer, especially those whose tumors carry less common mutations in a gene called BRAF, today’s targeted medicines simply do not work. This study introduces a new experimental drug, exarafenib, designed to hit a broader set of BRAF-driven tumors and explains how cancer cells try to escape it—revealing combination treatments that could keep these tumors under control longer.
A hidden majority of overlooked mutations
The researchers began by asking a basic question: how common are the different types of BRAF mutations in advanced cancers, and especially in non-small cell lung cancer (NSCLC)? Using a very large liquid-biopsy database of over 160,000 patients, they analyzed fragments of tumor DNA circulating in the bloodstream. They found that in lung cancer, BRAF mutations occur in roughly 4–5% of patients, which still translates into many thousands of people worldwide. Crucially, about two-thirds of these lung tumors carried so‑called Class II and Class III BRAF mutations—types that current approved BRAF drugs do not effectively target. Patients with one of these classes, especially Class II, tended to live for a shorter time than those with the more familiar Class I mutation, likely because Class I patients have access to established targeted therapies while Class II/III patients mostly receive standard chemotherapy or immunotherapy.
A next-generation drug that targets the full pathway
BRAF is part of a relay of proteins (the MAPK pathway) that passes growth signals from the cell surface into the nucleus. Many existing BRAF drugs were engineered to block only one mutant form and can even inadvertently stimulate related proteins in normal cells. Exarafenib was built differently: it is a “pan‑RAF” inhibitor designed to shut down several RAF family members (ARAF, BRAF, and CRAF) in both single and paired forms, while sparing most other cellular enzymes. In biochemical tests across hundreds of human kinases, exarafenib strongly blocked all three RAF proteins but had few off‑target effects, suggesting a cleaner safety profile than earlier pan‑RAF compounds.
Powerful effects in models of hard-to-treat tumors
The team tested exarafenib in a battery of cell lines and mouse models that carry different BRAF and RAS mutations, including realistic patient-derived tumors. In cell cultures, exarafenib curbed growth and shut down MAPK signaling not only in classic BRAF V600E cancer cells but also in those with Class II and Class III mutations and in many RAS-mutant cells that currently lack good targeted options. In mice bearing lung tumors with these alterations, exarafenib shrank or slowed tumors in a dose‑dependent fashion and showed clear relationships between drug levels, pathway suppression, and tumor response. Early clinical data from two patients with advanced BRAF‑mutant lung cancer—one with a rare BRAF fusion and another with a Class II point mutation—showed partial responses and meaningful symptom relief, supporting the relevance of the preclinical work.
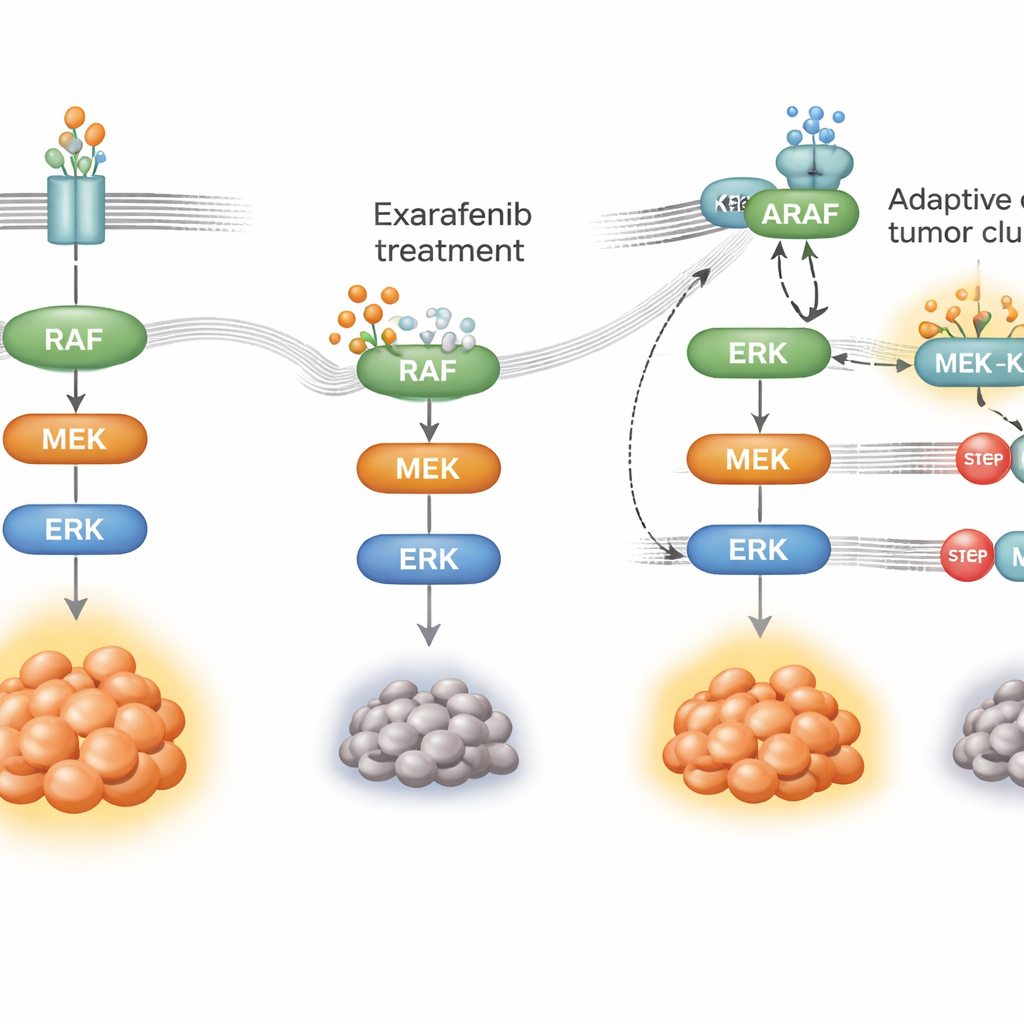
How cancer learns to bypass the drug
No targeted therapy works forever; cancer cells adapt. To see how tumors might resist exarafenib, the researchers exposed BRAF‑mutant lung cancer cells to the drug for months until resistant populations emerged. These cells still relied on the same growth pathway but had rewired how they used it. Instead of depending mainly on mutant BRAF, they ramped up activity of an upstream switch called RAS and shifted toward using another family member, ARAF, together with a scaffold protein named KSR1. Under drug pressure, ARAF and KSR1 formed tight complexes that helped keep the MAPK relay running even while exarafenib was bound. When the scientists selectively silenced ARAF or KSR1, or reduced RAS activity, the resistant cells once again became sensitive to exarafenib, and the survival signals collapsed.
Combination strategies to stay ahead of resistance
Armed with this mechanistic map, the team searched for partner drugs that could block the pathway at choke points shared by both the original and bypass routes. They found that pairing exarafenib with drugs that inhibit MEK or ERK—key downstream steps in the MAPK relay—produced strong synergy across many cell and mouse models, including tumors that were intrinsically less sensitive or had acquired resistance. These combinations kept the pathway shut longer, triggered more cell death, and in animals often worked as well as or better than higher doses of exarafenib alone, without obvious added toxicity. Agents that directly target RAS also enhanced exarafenib’s effects in models where RAS was clearly driving resistance, suggesting another clinically promising tactic.
What this means for patients
For people with NSCLC carrying Class II or Class III BRAF mutations—or complex BRAF fusions and RAS co‑mutations—there are currently no approved targeted therapies, and outcomes lag behind those of patients with more common alterations. This study provides a strong scientific case that exarafenib could help fill that gap by broadly shutting down RAF‑driven signaling. It also explains how tumors may adapt through an ARAF‑KSR1 bypass and shows that hitting the pathway at multiple levels, especially by combining RAF and MEK inhibitors or adding RAS inhibitors, could deliver deeper and more durable tumor control. Together, these insights chart a path toward clinical trials aimed at bringing tailored, combination targeted therapies to a large and previously underserved group of lung cancer patients.
Citation: Manabe, T., Bergo, H.C., Li, Q. et al. Pan-RAF inhibitor exarafenib targets BRAF class II/III NSCLC and reveals ARAF-KSR1 resistance and combination strategies. Nat Commun 17, 2484 (2026). https://doi.org/10.1038/s41467-026-69216-3
Keywords: BRAF-mutant lung cancer, pan-RAF inhibitor, MAPK signaling, drug resistance, targeted therapy combinations