Clear Sky Science · en
14-3-3ζ interacts with DNA-binding domain of FOXO3a and competitively dissociates DNA by dual-motif tethering
How Cancer Cells Shut Down Their Own Self-Destruct Switch
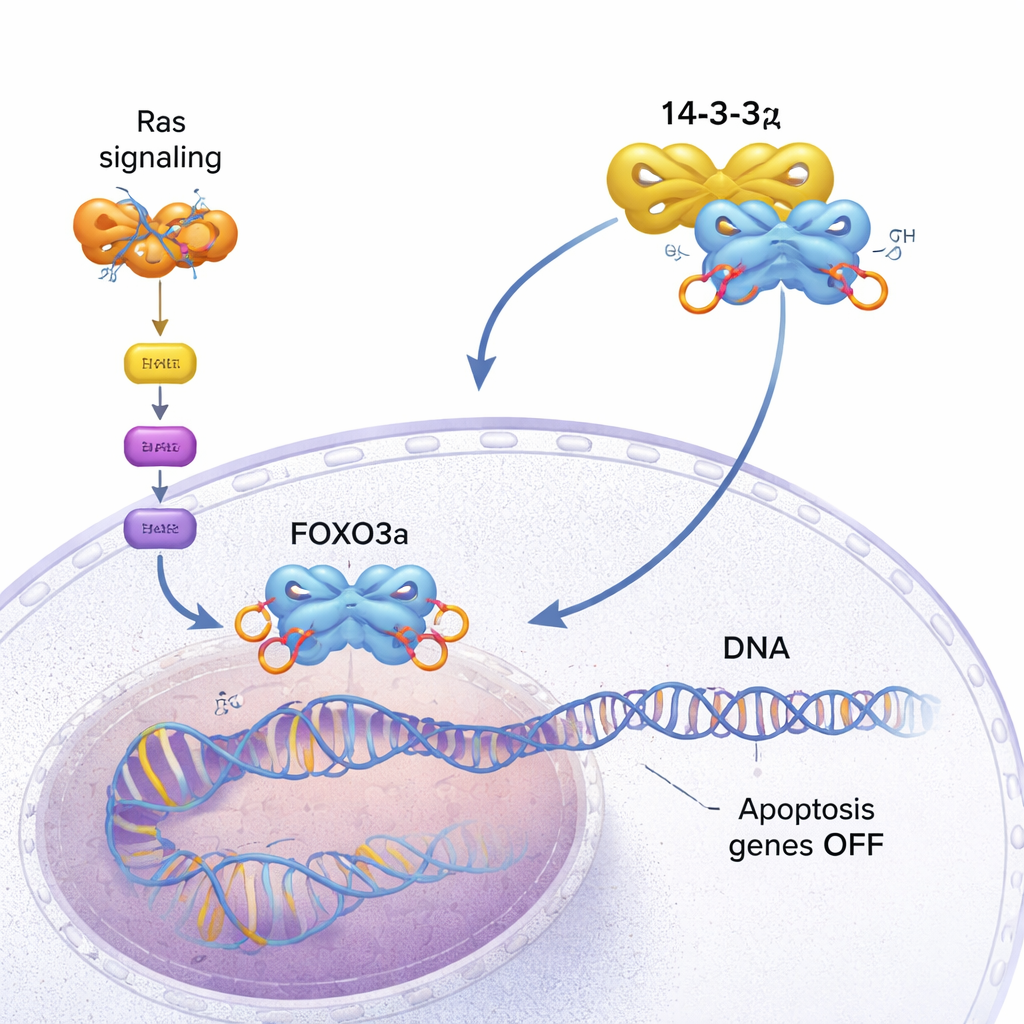
Our cells carry built‑in fail‑safe systems that can trigger their own death when they become too damaged or dangerous. One of these systems is controlled by a protein called FOXO3a, which turns on genes that make rogue cells commit suicide. Many cancers, however, find ways to silence FOXO3a and keep growing. This study digs into the molecular details of how another protein, 14‑3‑3ζ, helps cancer cells pry FOXO3a off DNA and disable this self‑destruct switch.
The Guardian Protein That Pushes Back Against Cancer
FOXO3a acts like a safety inspector for the cell. It binds to specific spots on DNA and activates genes that slow growth or trigger programmed cell death (apoptosis) when things go wrong. In healthy cells, this helps prevent tumors from forming. In many cancers, though, a growth‑promoting pathway driven by mutant Ras proteins becomes permanently switched on. This pathway activates a kinase called AKT, which chemically tags FOXO3a at several sites with phosphate groups. Those tags create docking spots for 14‑3‑3ζ, a dimeric “adapter” protein that recognizes phosphorylated motifs on many targets. When 14‑3‑3ζ latches onto FOXO3a, the cell’s internal brakes begin to fail.
Why Simple Binding Strength Couldn’t Explain the Effect
Earlier work on a related protein, FOXO4, suggested that 14‑3‑3 proteins pull FOXO factors off DNA simply because they bind more tightly. But FOXO3a prefers its natural DNA targets more strongly than the older model assumed. In this study, the researchers produced a version of FOXO3a that includes the DNA‑binding domain and two key phosphorylation sites. They measured how tightly this protein binds either DNA or 14‑3‑3ζ and found that the differences in binding strength were modest: 14‑3‑3ζ was only about twice as strong a partner as DNA. Yet in mixture experiments that track how molecules travel through a chromatography column, 14‑3‑3ζ was able to drive almost complete release of DNA from FOXO3a, as if it were roughly 100 times more competitive than expected. This mismatch hinted that an extra mechanism was at work.
A Three‑Point Grip That Crowds Out DNA
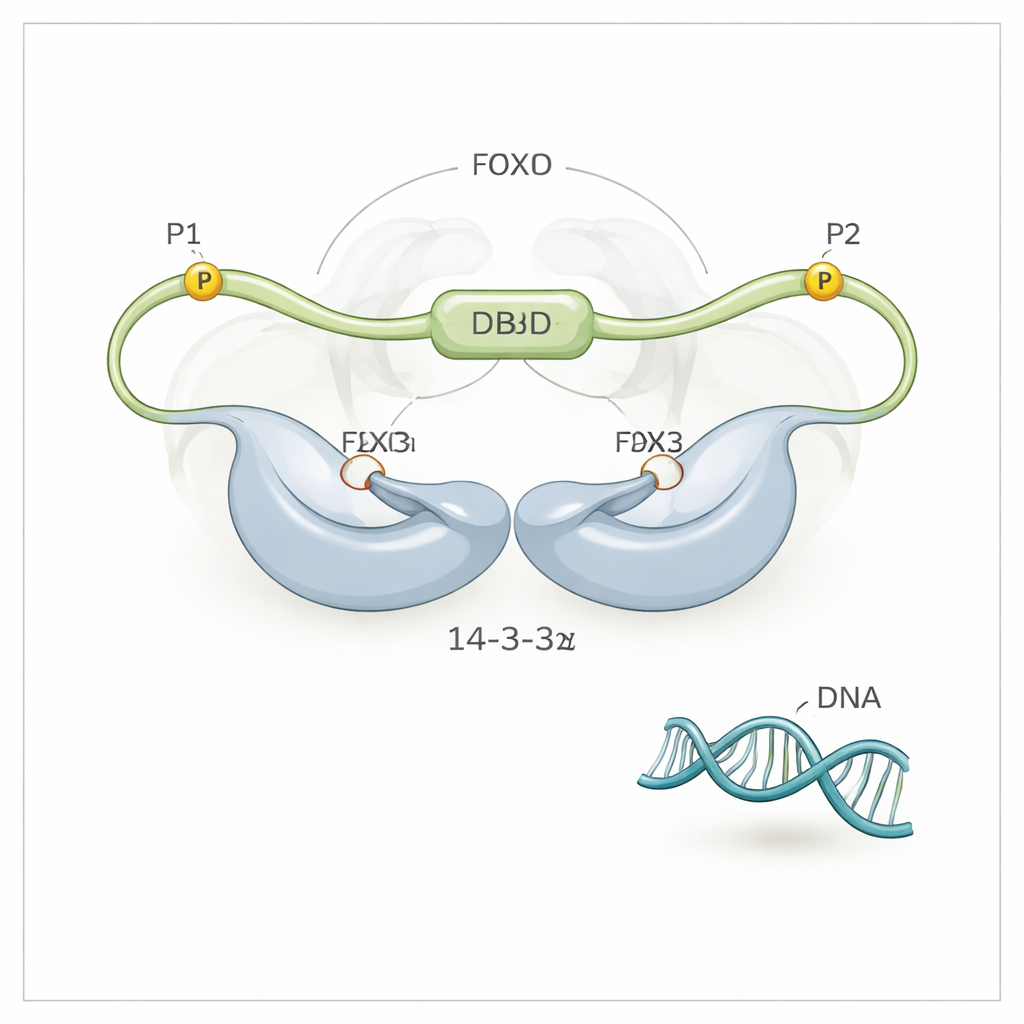
Using high‑resolution NMR spectroscopy, the team discovered that 14‑3‑3ζ does more than just grab FOXO3a at its two phosphorylated motifs (called P1 and P2). It also makes direct, though weaker, contact with FOXO3a’s DNA‑binding domain (DBD) itself—the very surface that normally hugs DNA. The 14‑3‑3ζ protein forms a symmetric dimer with two grooves. Each groove clamps onto one of the phosphorylated motifs on FOXO3a, tethering FOXO3a to 14‑3‑3ζ at two points. Because one of these motifs (P2) is very close along the chain to the DNA‑binding domain, this “dual‑motif tethering” effectively holds 14‑3‑3ζ right next to the DBD, greatly boosting the chance that the DBD will bump into and bind the surface of 14‑3‑3ζ instead of DNA. The researchers could even see that the DBD flips back and forth between the two sides of the 14‑3‑3ζ dimer, spending most of its time shielded from DNA.
Which Phosphate Tags Matter Most
To tease apart the roles of the two phosphorylated sites, the team engineered FOXO3a variants in which only one site could be phosphorylated at a time. When only the P2 site near the DBD was active, 14‑3‑3ζ could partially dislodge DNA but not completely. When only the more distant P1 site was active, 14‑3‑3ζ could bind FOXO3a but barely affected its grip on DNA. Full DNA release required both sites working together: P1 provides a high‑affinity initial docking point for 14‑3‑3ζ, and P2 positions the dimer close enough to the DBD to make the local concentration of 14‑3‑3ζ effectively enormous at that spot. This multistep tethering amplifies a modest binding preference into a powerful ability to evict DNA.
From Molecular Tug‑of‑War to New Drug Ideas
For a non‑specialist, the key takeaway is that cancer cells exploit a clever bit of molecular geometry, not just brute‑force binding strength, to silence a major tumor‑suppressing protein. 14‑3‑3ζ uses two small docking tags on FOXO3a as anchor points, then reaches over to cover the DNA‑gripping surface of FOXO3a’s core domain, preventing it from turning on cell‑death genes. Because the same FOXO and 14‑3‑3 families are widely used in many tissues, this dual‑tethering strategy is likely common in other cancers as well. Disrupting either the phosphate‑dependent anchors or the weaker contact with the DNA‑binding face of FOXO3a could restore its ability to activate self‑destruct programs in tumor cells, offering promising new angles for anticancer drug design.
Citation: Enomoto, S., Kuwayama, T., Nakatsuka, S. et al. 14-3-3ζ interacts with DNA-binding domain of FOXO3a and competitively dissociates DNA by dual-motif tethering. Nat Commun 17, 1503 (2026). https://doi.org/10.1038/s41467-026-69203-8
Keywords: FOXO3a, 14-3-3 proteins, apoptosis, Ras–AKT signaling, cancer therapeutics