Clear Sky Science · en
An equivariant pretrained transformer for unified 3D molecular representation learning
Teaching Computers to See Molecules in 3D
Designing new medicines and materials depends on understanding how molecules really look and move in three dimensions, not just as flat formulas on paper. This paper introduces a powerful new artificial intelligence model that can learn from the 3D shapes of many kinds of molecules at once — from small drug-like compounds to large proteins and their complexes — and then use that knowledge to predict how strongly they interact and which ones might become future drugs.
A Single Map for Many Molecular Worlds
Most current AI tools for chemistry are specialists: one is trained only on small molecules, another only on proteins, and a third only on their complexes. This separation wastes data and makes it hard to transfer what is learned in one area to another. The authors instead build a single “foundation” model, called the Equivariant Pretrained Transformer (EPT), that learns from a vast collection of 3D molecular structures drawn from multiple public databases. By treating all these structures within one shared framework, the model can recognize common patterns in how atoms arrange and interact, whether they belong to a simple drug molecule or to a complex knot of protein chains. 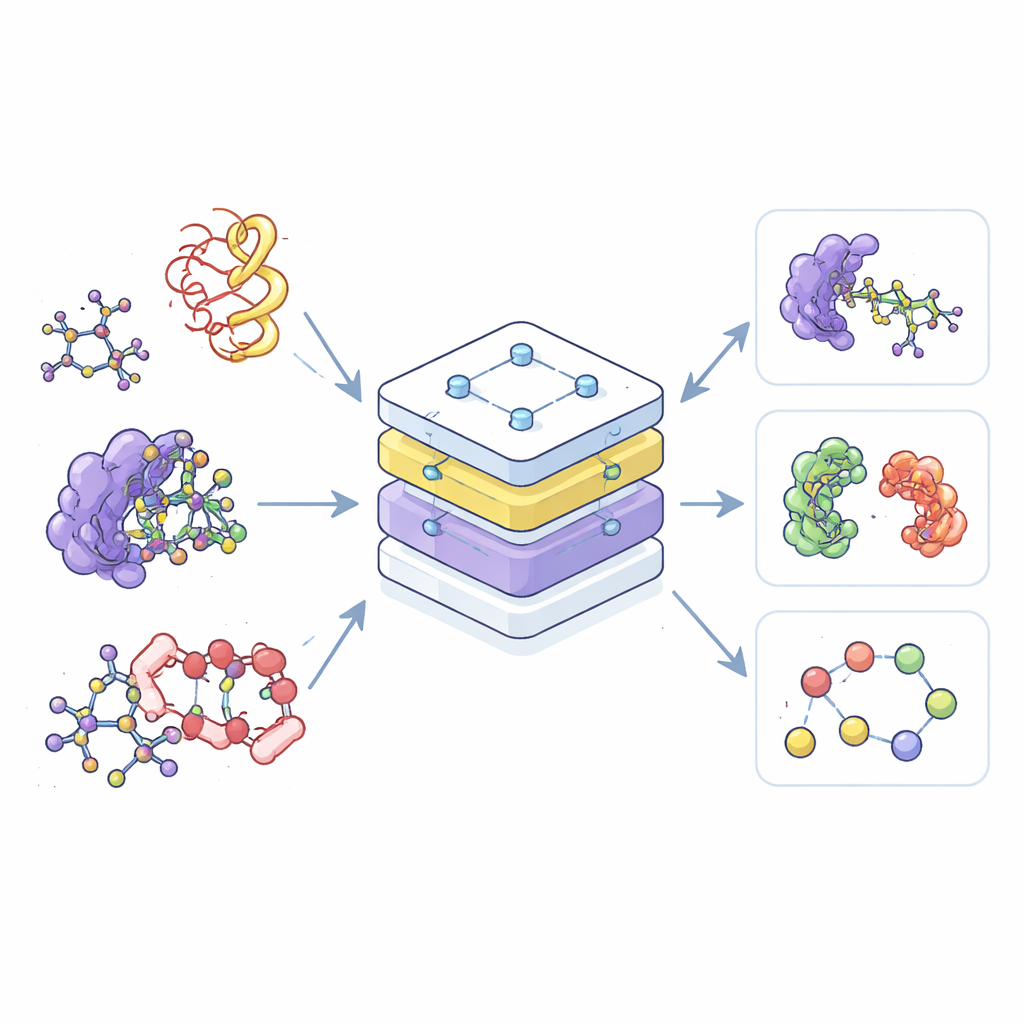
Breaking Molecules into Manageable Pieces
To cope with the huge variety and size of molecular systems, the researchers introduce the idea of “blocks” — small, meaningful chunks of atoms. For small molecules, a block groups a heavy atom with its attached hydrogens; for proteins, each amino acid becomes a block. During training, the model sees both the fine-grained atoms and the coarser block structure, allowing it to connect local chemical details with broader 3D shapes such as protein backbones or binding pockets. This block view also creates a common language that works across very different molecular types, making it possible for one model to understand them all.
Learning by Cleaning Up Noisy Structures
Instead of being told explicit labels like “this molecule is soluble” or “this one binds tightly,” EPT is trained in a self-supervised way. The authors deliberately disturb each molecular block, randomly shifting and rotating it away from its true position, and then ask the model to infer the forces and twists needed to restore the original structure. Because the training respects fundamental geometric rules — the molecule should look the same if the entire system is rotated or moved in space — the model learns a physically sensible sense of 3D shape. This denoising game teaches EPT how atoms within and between blocks hold together and how subtle changes in geometry affect stability. 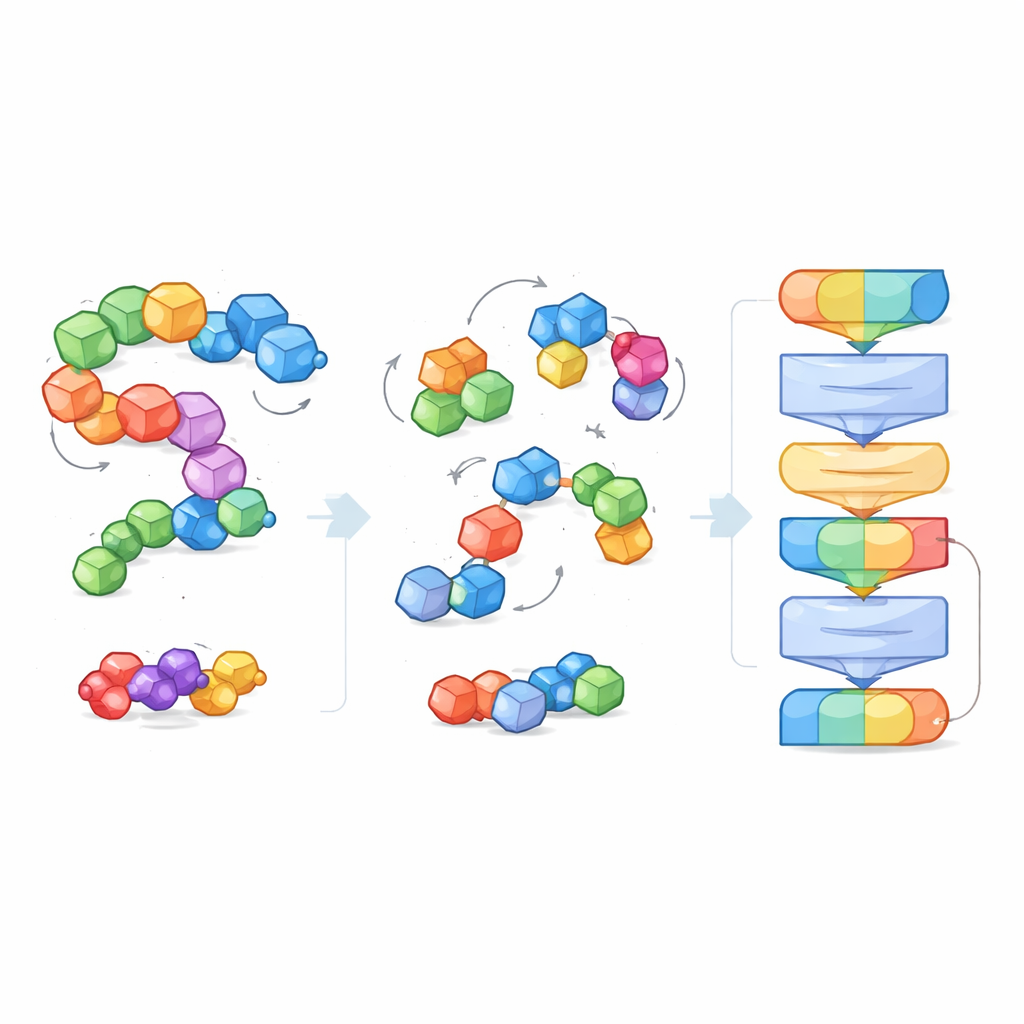
Putting the Model to the Test
After pretraining on more than five million structures, EPT is fine-tuned for several real scientific tasks. It predicts how strongly a small molecule binds to a protein pocket, how a single mutation at a protein interface affects binding, and key physical properties of small molecules that chemists care about. Across diverse benchmarks, the unified model matches or outperforms the best existing tools, including those that were carefully tailored for only one domain. Notably, when trained on one type of data, such as small molecules, it still helps with seemingly different tasks, like protein binding, revealing that it has captured broadly useful chemical principles rather than narrow tricks.
Searching for New COVID‑19 Treatments
The authors further demonstrate EPT’s practical value by turning it loose on a drug repurposing challenge. They first fine-tune the model on protein–ligand complexes and then use it to rank nearly 2,000 already approved drugs by their predicted ability to bind the main protease of SARS‑CoV‑2, a key enzyme the virus needs to replicate. Known anti‑COVID‑19 drugs rise toward the top of the ranking, and the model highlights additional promising candidates. Twelve top-scoring molecules are examined more closely with computer simulations, and two — including one not originally developed for COVID‑19 — show especially strong predicted binding and are confirmed experimentally to inhibit the viral protease at micromolar levels.
A Step Toward General Molecular AI
In plain terms, this work shows that a single, geometry-aware AI model can learn a shared 3D understanding of many molecular systems and then use it to answer a wide range of scientific questions. By organizing molecules into blocks and training the model to “repair” distorted structures, the authors create a tool that not only predicts numbers more accurately but can also accelerate tasks like finding new antiviral drugs. EPT points toward a future where general-purpose molecular AI systems help chemists and biologists explore chemical space more efficiently, guiding experiments and shortening the path from atomic structure to practical therapies and materials.
Citation: Jiao, R., Kong, X., Zhang, L. et al. An equivariant pretrained transformer for unified 3D molecular representation learning. Nat Commun 17, 2606 (2026). https://doi.org/10.1038/s41467-026-69185-7
Keywords: 3D molecular representation, equivariant transformer, drug discovery, protein–ligand binding, self-supervised learning