Clear Sky Science · en
Molecular determinant of low-voltage dependence of human Nav1.7 inactivation revealed by efficacy-based Nav1.7 selective inhibitor
Turning Down the Volume on Pain Signals
Why do some people feel excruciating pain from a light touch, while others hardly feel pain at all? A big part of the answer lies in tiny protein gateways in our nerve cells that control electrical signals. This study uncovers how a subtle structural quirk in one such gateway, called Nav1.7, makes it especially important for triggering pain — and how a natural compound, Uvarigranol D, can selectively shut it down. The work points toward a new strategy for designing painkillers that quiet overactive pain nerves without slowing the heart or clouding the brain. 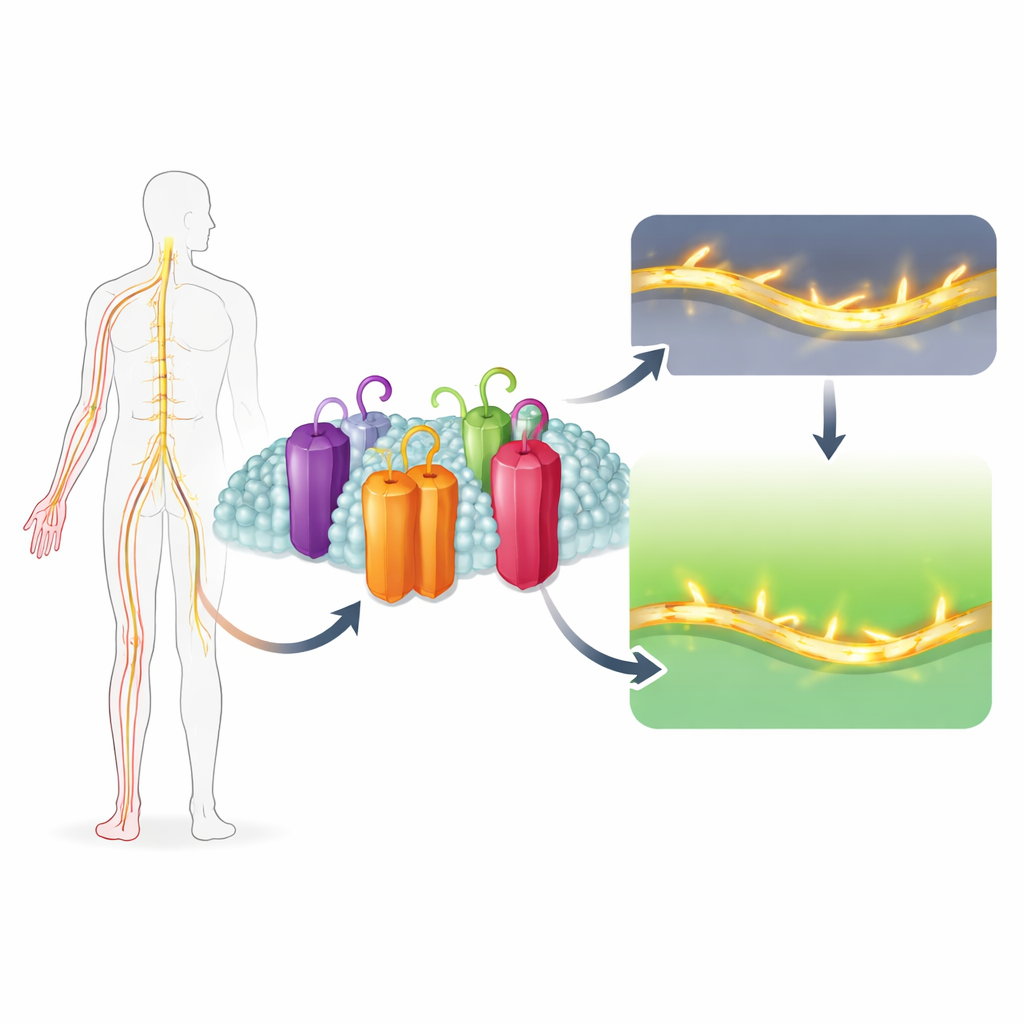
A Special Gatekeeper for Pain
Our nerves fire electrical impulses using sodium channels, microscopic pores that briefly open to let positively charged sodium ions rush into the cell. There are nine main versions of these channels in humans, each tuned to different tissues such as brain, muscle, heart, or pain-sensing nerves. Nav1.7 is the version packed into peripheral pain fibers. It is unusual because it can turn on and off at lower voltages than its relatives, allowing it to respond to even tiny nudges in voltage. This makes Nav1.7 a powerful amplifier of weak, pain-triggering signals. Genetic studies show that overactive Nav1.7 causes severe inherited pain syndromes, while completely nonfunctional Nav1.7 makes people unable to feel pain at all.
Finding a Pain-Selective Blocker
Drug developers have long hoped to target Nav1.7 to treat chronic pain, but it closely resembles other sodium channels that are vital for heartbeat and brain function. Most experimental drugs latch onto many channel types, causing side effects or failing in clinical trials. The researchers screened over 1,500 natural compounds using a cell-based assay that detects changes in membrane voltage. They identified a family of molecules from the plant Uvaria grandiflora, focusing on one called Uvarigranol D (UGD). UGD dampened sodium currents in several channel types, but it almost completely silenced Nav1.7 while only half-blocking other sodium channels even at high doses. This means its selectivity comes not from tighter binding, but from a much stronger effect once it binds. 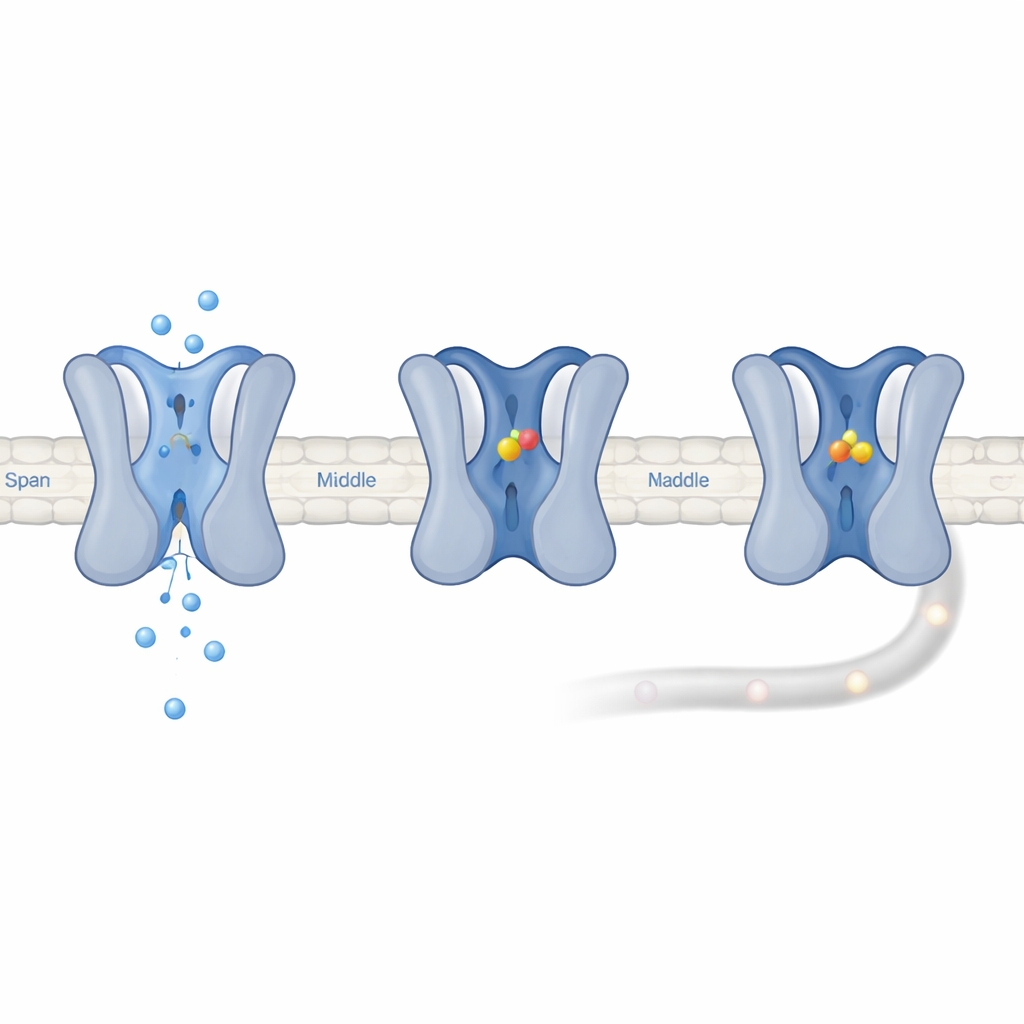
A Single Atom-Sized Change Makes Nav1.7 Unique
To understand why UGD is so effective on Nav1.7, the team built chimeric channels that swapped pieces between Nav1.7 and a closely related brain channel, Nav1.2. This pinpointed a small region near the outer mouth of the pore, between two structural segments called S5 and S6 in domain III, as the key determinant of UGD’s full blocking power. Comparing amino acid sequences revealed that Nav1.7 alone carries a threonine at position 1398, whereas all other human sodium channels carry a bulkier methionine there. When the researchers replaced Nav1.7’s threonine with methionine, UGD could no longer fully shut the channel; swapping methionine for threonine in Nav1.2 made that channel behave like Nav1.7. This single substitution also shifted when the channels turned on and off with voltage: threonine made the channel open and inactivate at more negative voltages and do so more quickly, properties that favor Nav1.7 being in a non-resting state even near the normal resting voltage of pain neurons.
Locking the Channel in a Resting-Down Mode
Electrical recordings showed that UGD does not stick to Nav1.7 when it is closed or briefly open. Instead, it prefers channels that have entered a long-lasting “slow inactivated” state, in which the pore is closed and takes hundreds of milliseconds or longer to recover. When UGD was present, the channels took about ten times longer to escape from this state, meaning UGD stabilizes it. Computer simulations suggested that UGD nestles into a pocket formed where one loop of domain III meets a helix in domain IV, making key contacts with five amino acids. Mutating any of these residues weakened UGD’s effect, confirming the pocket’s importance. Because Nav1.7’s threonine-rich structure makes it slide into inactivated states at lower voltages, more of its channels are in the very state that UGD prefers to bind, explaining why Nav1.7 is functionally much more suppressed than its cousins even though their binding strength is similar.
From Ion Pores to Pain Relief
Ultimately, what matters is how these molecular events influence real cells. In rat pain-sensing neurons from the dorsal root ganglion, UGD greatly reduced the number of action potentials — the rapid spikes of voltage that carry pain information — and eventually stopped them altogether at low micromolar and submicromolar concentrations. In contrast, human heart-like cells derived from stem cells, which mainly use a different sodium channel (Nav1.5) and rest at a slightly lower voltage, were about 60 times less sensitive. This suggests that an “efficacy-selective” blocker like UGD can dampen pain pathways far more than cardiac or other excitable tissues simply because of how often Nav1.7 sits in its inactivated, drug-favored state.
What This Means for Future Pain Treatments
The study reveals that a small structural detail — a single threonine — underlies Nav1.7’s special low-voltage behavior and its ability to generate “threshold currents,” the tiny signals that decide whether a pain neuron will fire. By binding and stabilizing the inactivated form of this channel, UGD exploits that built-in tendency and suppresses Nav1.7 much more strongly than other sodium channels. For a layperson, the takeaway is that the authors have mapped a precise weak spot in the body’s pain gatekeeper and shown a way to press on it without heavily disturbing heart or brain channels. This insight opens a path to designing new painkillers that quiet overactive pain nerves by targeting voltage behavior and channel state, rather than just chasing tighter binding to Nav1.7.
Citation: Zhao, F., Xi, C., Li, J. et al. Molecular determinant of low-voltage dependence of human Nav1.7 inactivation revealed by efficacy-based Nav1.7 selective inhibitor. Nat Commun 17, 2559 (2026). https://doi.org/10.1038/s41467-026-69184-8
Keywords: Nav1.7 sodium channel, chronic pain, Uvarigranol D, state-dependent inhibition, voltage-gated sodium channels