Clear Sky Science · en
Atomic resolution ensembles of intrinsically disordered proteins with Alphafold
Why shape-shifting proteins matter
Our cells are full of proteins that never settle into a single, rigid shape. These "intrinsically disordered" proteins behave more like floppy noodles than neatly folded machines, yet they are central to processes ranging from cell signaling to neurodegenerative disease. Because they constantly move and flex, capturing their full range of shapes at atomic detail is extremely hard and usually requires years of experiments and heavy computations. This article presents a new way to harness artificial intelligence and physics together to map these restless molecules far more efficiently.
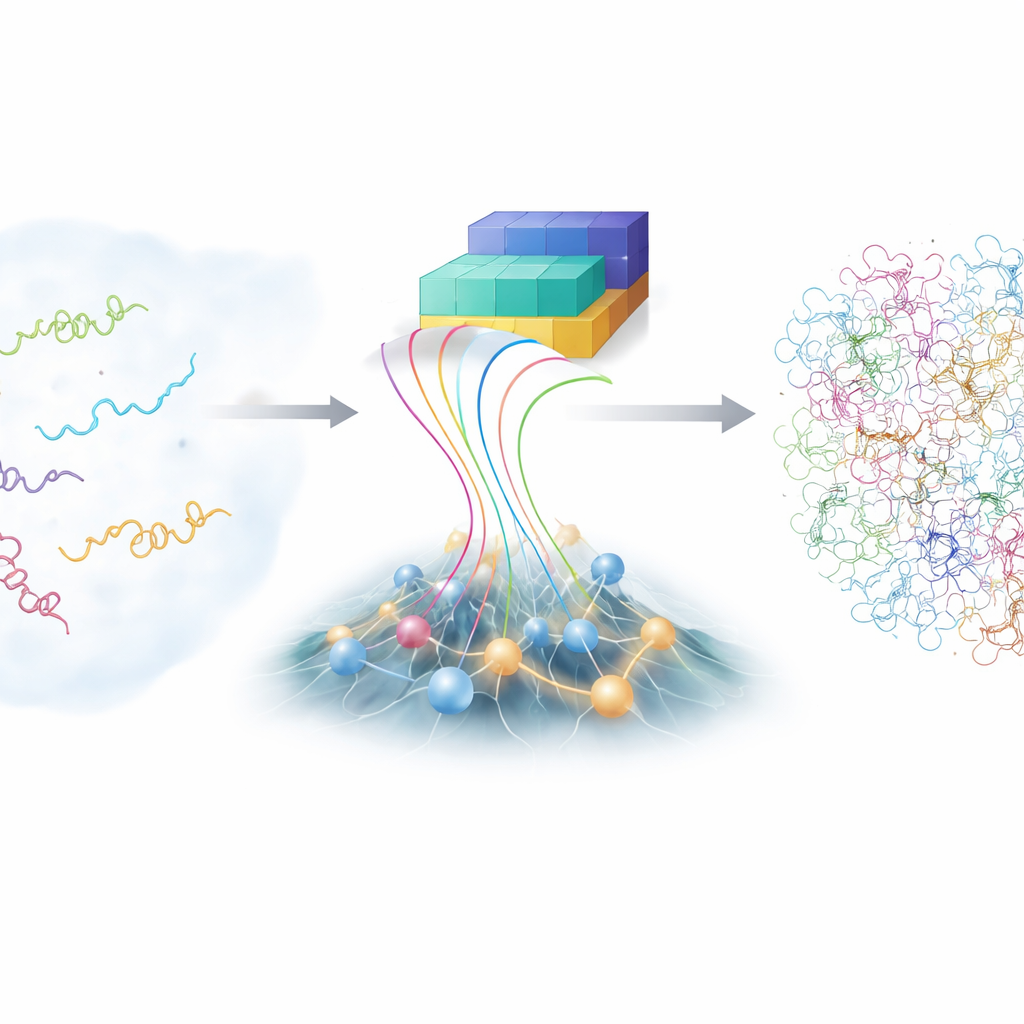
The challenge of restless molecules
Unlike textbook protein models that show one tidy structure, intrinsically disordered proteins (IDPs) roam through a vast landscape of possible shapes. That flexibility helps them recognize many different partners, but it also makes them notoriously difficult to study. Traditional laboratory techniques, like advanced nuclear magnetic resonance and X-ray scattering, can report on averages over many shapes but not on each individual form. Computer simulations at full atomic detail can, in principle, follow every atom as an IDP wriggles, yet they are extremely expensive and depend on finely tuned physical models. As a result, the scientific community has only a limited collection of accurate, detailed IDP ensembles to learn from.
Combining smart guesses with physical rules
In recent years, the AlphaFold family of deep learning tools has stunned biology by predicting protein structures from their amino-acid sequences. For disordered proteins, however, AlphaFold’s usual strength—guessing a single best shape—is less helpful, because IDPs do not have just one. What AlphaFold does provide, though, is rich information about how likely different parts of the chain are to be near or far from each other. The authors built a new framework, called bAIes, that treats this AI-derived information as soft guidance and blends it with a fast, physics-based model that deliberately starts from a "random coil" view, where the chain explores all possible bends and twists without favoring any particular structure.
From random tangles to realistic ensembles
First, the researchers constructed an efficient physical model that reproduces how a completely unstructured protein chain behaves, based on statistics extracted from thousands of known protein structures. This model serves as the "prior"—the baseline expectation of how an IDP moves if we know nothing else. Next, bAIes reads AlphaFold’s predictions about which residue pairs tend to come close. Rather than forcing the protein into a single pattern, it converts those hints into gentle distance restraints with built-in uncertainty, allowing the chain to satisfy the AI’s suggestions only when they are consistent with the broader physical picture.
Testing against real experiments
To see whether this approach works, the team applied bAIes to a set of 21 proteins ranging from almost fully random coils to more complex systems with transient helices and multiple domains. For each, they compared the resulting computer-generated ensembles to a wide array of experimental measurements that probe both local details and global size and shape. For very floppy proteins such as the Alzheimer’s-related peptide Aβ40, the simple random coil model was already close to reality, and bAIes preserved this good agreement. For partially structured proteins, bAIes improved the match to experiments by correctly capturing where short helical segments and compact patches appear and disappear. Crucially, the method stayed robust even when AlphaFold was overconfident and mistakenly predicted stable folds where solution experiments show disorder, because bAIes explicitly allows for errors in the AI input.
Beating or matching existing methods
The authors then pitted bAIes against heavyweight all-atom simulations run on specialized supercomputers, leading coarse-grained models that simplify proteins into beads, and new deep learning generators trained on simulation data. Across multiple tests, bAIes consistently matched or outperformed these approaches in reproducing experimental data, while being far less computationally demanding than full-scale simulations. It also worked beyond simple IDPs, handling proteins with several rigid domains linked by flexible connectors and recovering their overall shapes in solution. When the researchers further fine-tuned the bAIes ensembles with experimental data, the agreement improved even more, showing that the method can serve as a powerful starting point for integrative modeling.
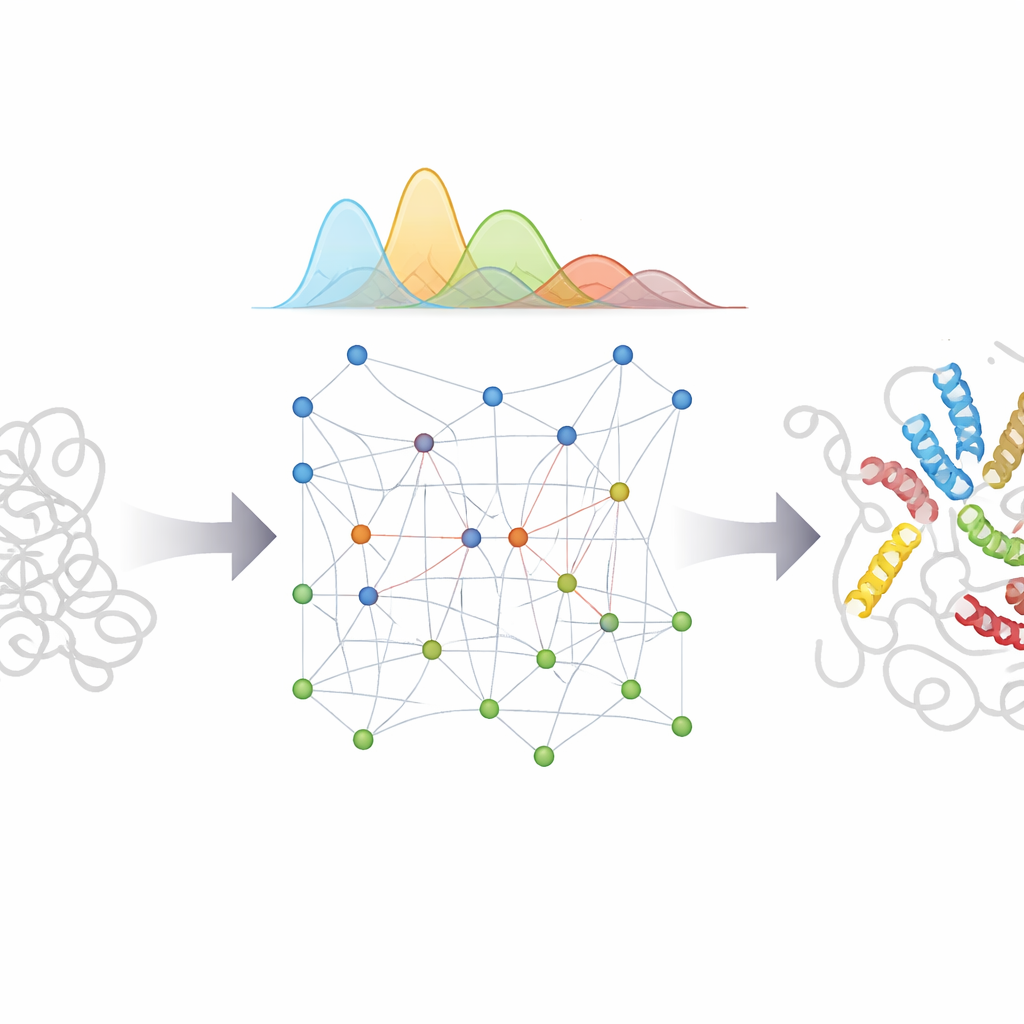
What this means for biology and medicine
By marrying AlphaFold’s pattern-recognition power with a carefully designed physical model and a Bayesian treatment of uncertainty, bAIes offers a practical route to detailed "movies" of disordered proteins rather than single snapshots. These atomically detailed ensembles can help scientists understand how flexible regions recognize partners, how misfolding and aggregation begin in diseases like Parkinson’s and Alzheimer’s, and how small molecules might bind to elusive, shifting targets. Because the method is efficient and built into open-source software, it can be widely adopted to generate realistic ensembles for many disordered proteins, guiding experiments and supporting future AI systems that aim to predict not just one structure, but the full range of shapes that life’s most flexible molecules can adopt.
Citation: Schnapka, V., Morozova, T.I., Sen, S. et al. Atomic resolution ensembles of intrinsically disordered proteins with Alphafold. Nat Commun 17, 2399 (2026). https://doi.org/10.1038/s41467-026-69172-y
Keywords: intrinsically disordered proteins, AlphaFold, Bayesian modeling, protein ensembles, structural biology