Clear Sky Science · en
Inhibition mechanism of the fungal β−1,3-glucan synthases by triterpenoid antifungal drugs
Why stopping fungal infections matters
Fungal infections quietly kill more than a million people every year and threaten crops and ecosystems worldwide. Doctors already struggle with a short list of antifungal medicines, and some dangerous fungi are evolving to evade them. This study uncovers, in molecular detail, how a new class of antifungal drugs latch onto and disable a key enzyme that fungi need to build their protective cell wall. Understanding this process offers a roadmap for designing better treatments that can outsmart resistant fungi.
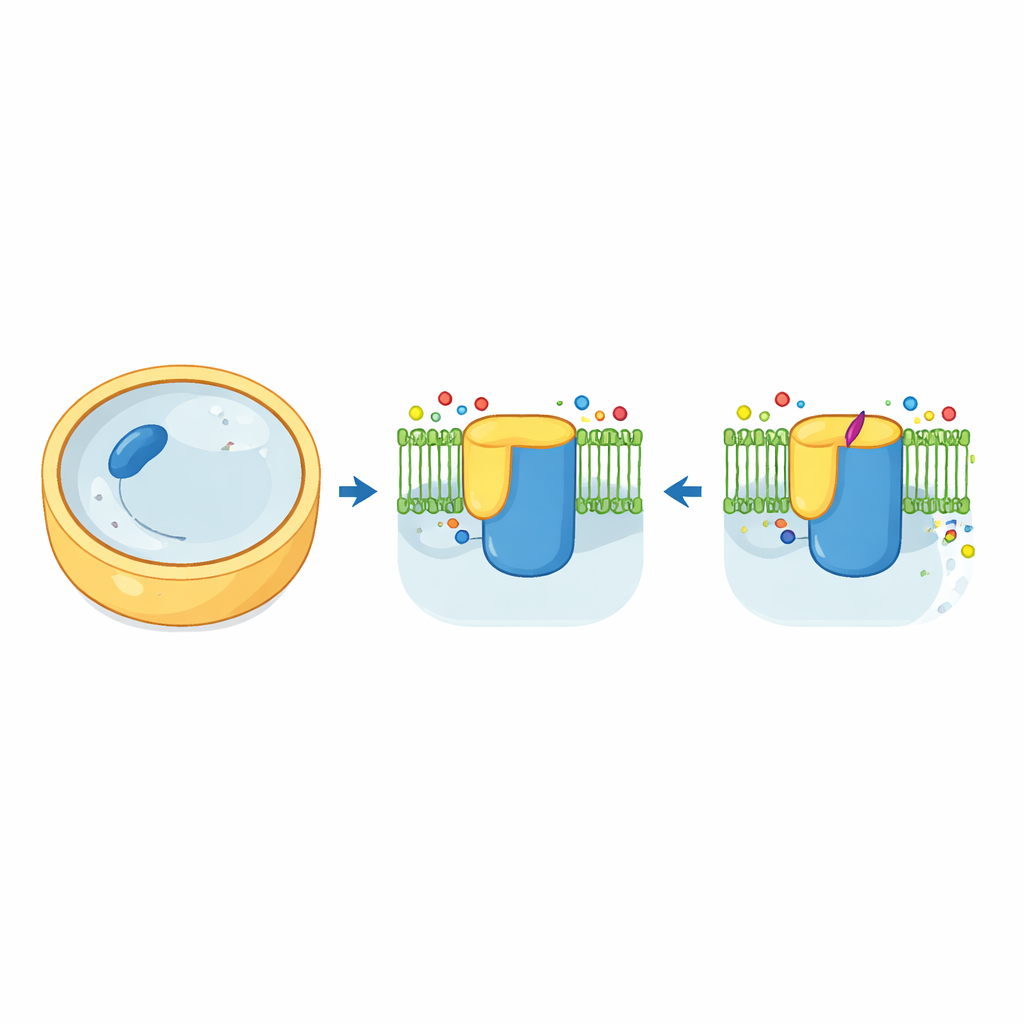
The brickmaker of the fungal cell wall
Fungal cells are wrapped in a tough wall made largely of a sugar-based polymer called beta-1,3-glucan. The enzyme that manufactures and exports this polymer, known as beta-1,3-glucan synthase, acts like a brickmaker and conveyor belt combined. In baker’s yeast, this machine is built from two very similar versions, Fks1 and Fks2, each embedded in the cell membrane and controlled by a small helper protein called Rho1. Using cryo-electron microscopy, the authors captured high-resolution three-dimensional views of both Fks1 and Fks2 in their natural form. The structures reveal a large cytosolic “workbench” joined to a forest of membrane-spanning helices, with a central cavity that likely serves as the tunnel through which the growing glucan chain exits the cell.
How a natural product drug grabs the enzyme
Clinically important triterpenoid drugs, including the oral medicine ibrexafungerp, trace their origins to a natural compound called enfumafungin. Until now, nobody knew exactly where on the glucan synthase these drugs bind. The researchers solved structures of both Fks1 and Fks2 bound to enfumafungin. Surprisingly, the drug does not wedge into the active site where sugar units are joined. Instead, it nestles on the outer portion of a single membrane helix called TM5, sitting in the membrane about three nanometers away from the catalytic center and near the likely glucan exit channel. Key amino acids in this patch cradle the hydrophobic core of the drug and contact its acidic tail, while its sugar appendage barely touches the protein—explaining why chemists have been able to modify that part without losing potency.
Drugs that freeze a moving machine
At first glance, the overall shape of the enzyme with and without enfumafungin looks nearly unchanged. The crucial difference lies in how the surrounding lipids are organized and how flexible certain helices are. When the drug binds, a nearby aromatic side chain rotates to clamp enfumafungin in place and, together with another residue, stabilizes an ordered lipid alongside the drug. Additional sterol-like lipids become neatly arranged around a set of horizontal helices that help define the path for glucan export. These lipids act like wedges and props, locking the helices and channel entrance into a particular “basal” arrangement. Genetic tests show that altering many of the contact points for the drug or these sterols weakens drug binding or destroys enzyme function, and mutations at these sites match known resistance mutations in human and plant pathogens.
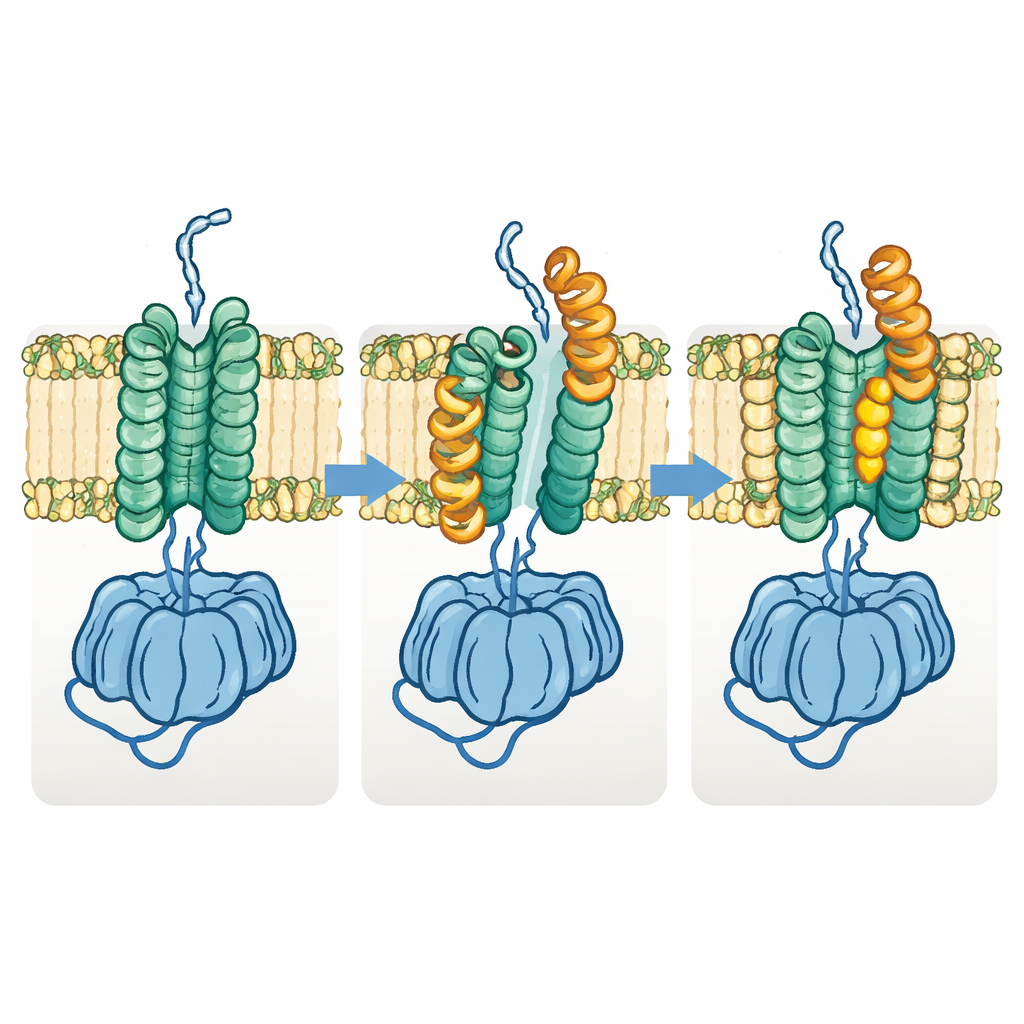
A necessary shape-shift for polymer transport
The team also discovered a distinct “open” form of Fks1. In this state, one half of the membrane region shifts sideways and key horizontal helices swing outward, prying open the space between the two membrane bundles and widening the apparent glucan tunnel. Comparing many structures suggests that the enzyme cycles between the basal and open states during normal operation: the basal state prepares and initiates polymer formation, while the open state allows the growing chain to move laterally through the membrane toward the outside. When the authors engineered disulfide links intended to lock the enzyme permanently in either state, both variants largely lost activity, supporting the idea that this shape-shifting is essential for function.
What this means for future antifungal drugs
By showing that enfumafungin and related drugs work not by blocking the catalytic pocket but by reshaping the local membrane environment and freezing the glucan synthase in its basal state, this study reveals an unconventional mode of drug action. It also demonstrates that Fks1 and Fks2 share nearly identical structures and drug responses, explaining why both must be considered when tackling resistance. More broadly, the work highlights how small molecules can control “undruggable” membrane proteins by binding shallow surfaces and recruiting lipids, offering a conceptual blueprint for designing next-generation antifungals that remain effective even as fungi evolve.
Citation: You, ZL., Sun, L., Wang, LX. et al. Inhibition mechanism of the fungal β−1,3-glucan synthases by triterpenoid antifungal drugs. Nat Commun 17, 2347 (2026). https://doi.org/10.1038/s41467-026-69114-8
Keywords: antifungal drugs, fungal cell wall, glucan synthase, drug resistance, cryo-electron microscopy