Clear Sky Science · en
scLong: a billion-parameter foundation model for capturing long-range gene context in single-cell transcriptomics
Teaching Computers to Read the Hidden Language of Cells
Every cell in your body carries a bustling city of genes turning on and off in intricate patterns. Modern single-cell RNA sequencing can now listen in on each individual cell, but the result is an overwhelming flood of numbers. This paper introduces scLong, a massive artificial intelligence model designed to make sense of these complex gene activity patterns, including faint signals that older methods tend to ignore. Its goal is to help researchers understand how cells react to genes being switched, drugs being added, or diseases taking hold.
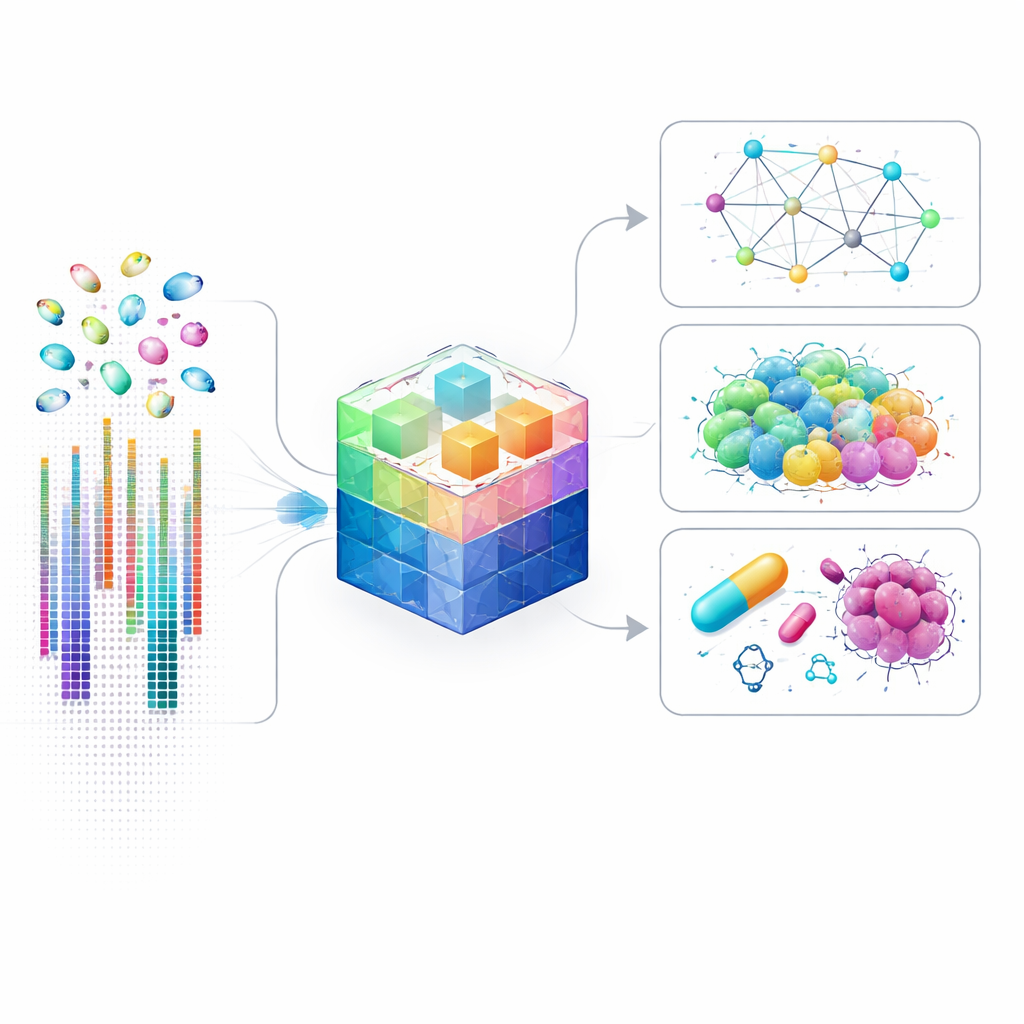
Why Cell-Level Gene Maps Matter
Traditional gene studies often mix together millions of cells, averaging out rare or unusual ones. Single-cell techniques changed that by measuring gene activity in each cell separately, revealing hidden cell types, subtle cell-to-cell communication, and detailed control circuits that decide what a cell will do. However, analyzing this kind of data is extremely challenging: each cell may have activity levels measured for tens of thousands of genes, many of which are barely detectable. Existing AI models simplify the problem by focusing only on the loudest genes, which speeds up computation but misses many subtle signals that might be crucial in disease, development, or drug response.
A New Model That Listens to Every Gene
scLong tackles this challenge by scaling up instead of trimming down. It is a billion-parameter foundation model trained on gene activity profiles from about 48 million human cells across more than 50 tissues. Unlike earlier approaches that attend to a few thousand highly active genes, scLong considers roughly 28,000 genes at once, including those that are rarely or weakly expressed. It combines two kinds of information for each gene: how active it is in a given cell and what is already known about its function from the Gene Ontology, a large expert-curated catalog of gene roles and relationships. A specialized network operating on a graph of gene connections distills this prior knowledge into compact representations that the model can use alongside the raw expression values.
How the Model Balances Power and Efficiency
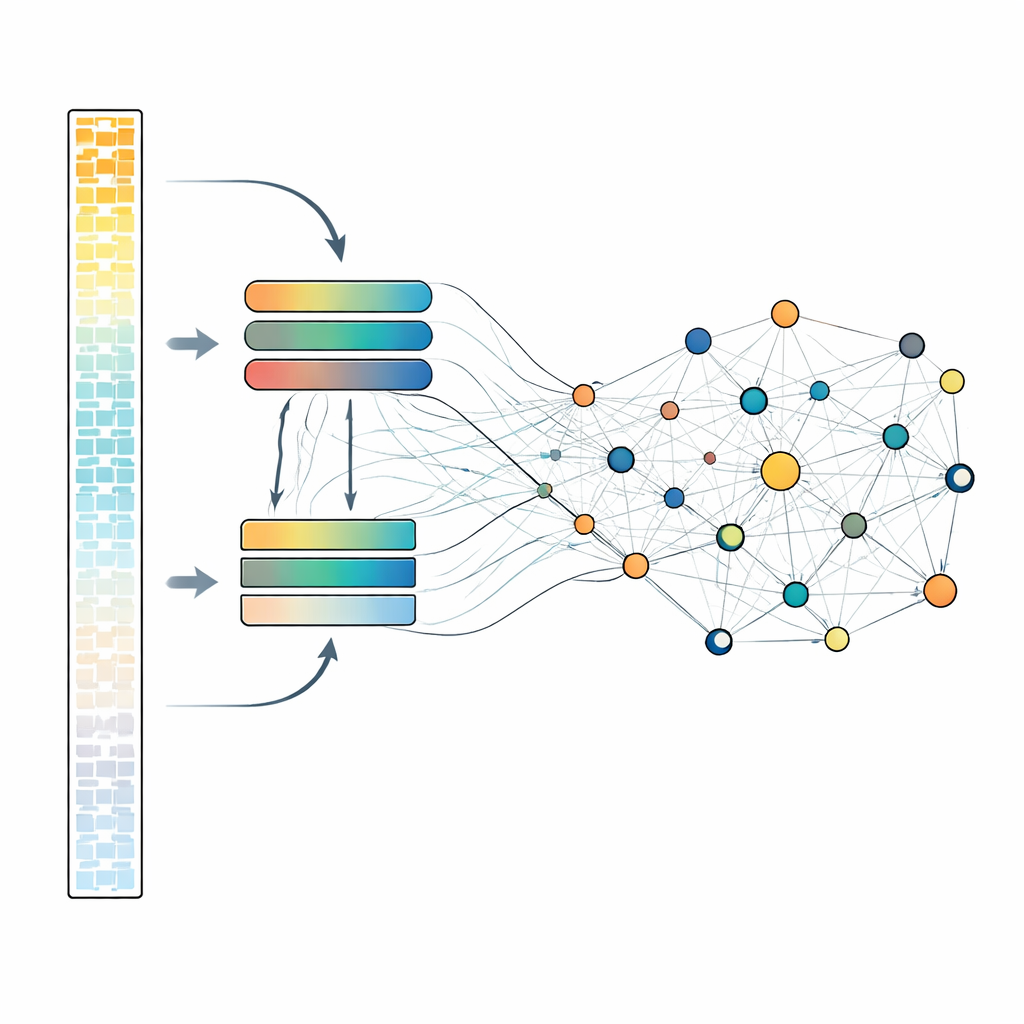
Looking at every gene in detail is computationally expensive, so scLong uses a clever two-track design. Within each cell, genes are sorted by how strongly they are expressed. The most active genes, which often carry the main biological signal, are processed through a larger, more powerful attention module. The quieter genes, including low and even zero measurements, are routed through a smaller, lighter-weight module. Afterwards, all genes are brought back together and passed through another attention layer that lets every gene influence every other. This design allows the model to keep cheaper but still meaningful representations for faint signals while reserving more capacity for the strongest ones. During pretraining, the system repeatedly hides a subset of gene activity values and learns to reconstruct them from the surrounding context, forcing it to discover the patterns that link genes together.
Putting the Model to Work on Real Problems
Once trained, scLong can be adapted to a wide range of biological questions. The authors show that it predicts how gene activity will change when specific genes are switched off or altered, including combinations of two genes that may act together. It also forecasts how cells respond when exposed to different chemicals, which is important for drug discovery and safety testing. In cancer studies, scLong helps anticipate how tumor cell lines will respond to single drugs and to pairs of drugs that might work better in combination, often outperforming both specialized models and other large foundation models. Beyond prediction, scLong can infer networks of regulatory relationships between genes and can help correct technical distortions that arise when data are collected in different laboratories or on different machines.
What This Means for Future Medicine and Research
In simple terms, scLong gives scientists a high-resolution, context-aware map of gene activity inside individual cells, one that does not throw away the quiet or rarely used genes. By learning from millions of cells and incorporating existing biological knowledge, it offers more accurate guesses about how cells will react when genes are disrupted, when new drugs are introduced, or when disease processes unfold. This could accelerate the search for new therapies, guide more personalized treatment choices, and sharpen our understanding of how complex gene networks control health and disease. While the model is large and computationally demanding, it points toward a future in which powerful, general-purpose AI systems serve as versatile companions for exploring the hidden workings of our cells.
Citation: Bai, D., Mo, S., Zhang, R. et al. scLong: a billion-parameter foundation model for capturing long-range gene context in single-cell transcriptomics. Nat Commun 17, 2380 (2026). https://doi.org/10.1038/s41467-026-69102-y
Keywords: single-cell transcriptomics, foundation models, gene regulation, drug response prediction, gene expression