Clear Sky Science · en
Cross ionization mode chemical similarity prediction between tandem mass spectra in metabolomics
Why connecting chemical dots matters
Every sip of coffee, breath of air, or course of medicine leaves behind tiny chemical traces in our bodies. Modern instruments can detect thousands of these molecules at once, but turning those signals into biological insight is still surprisingly hard. This study introduces MS2DeepScore 2.0, a machine-learning tool that helps scientists see how these molecules are related, even when the signals are recorded in very different ways. By doing so, it promises faster and more complete interpretations of complex chemical mixtures in medicine, nutrition, and environmental research.
Two ways of looking at the same molecule
Mass spectrometry is a workhorse technique that weighs and breaks apart molecules to reveal their identity. In routine experiments, scientists often measure the same sample in two modes: one that favors positively charged molecules and one that favors negatively charged ones. Each mode produces its own characteristic “barcode” of fragments. Even when both measurements come from the same molecule, the resulting patterns can look so different that traditional comparison methods fail. As a result, researchers usually analyze the two modes separately, build two disconnected maps of the sample, and risk missing important relationships between chemicals.
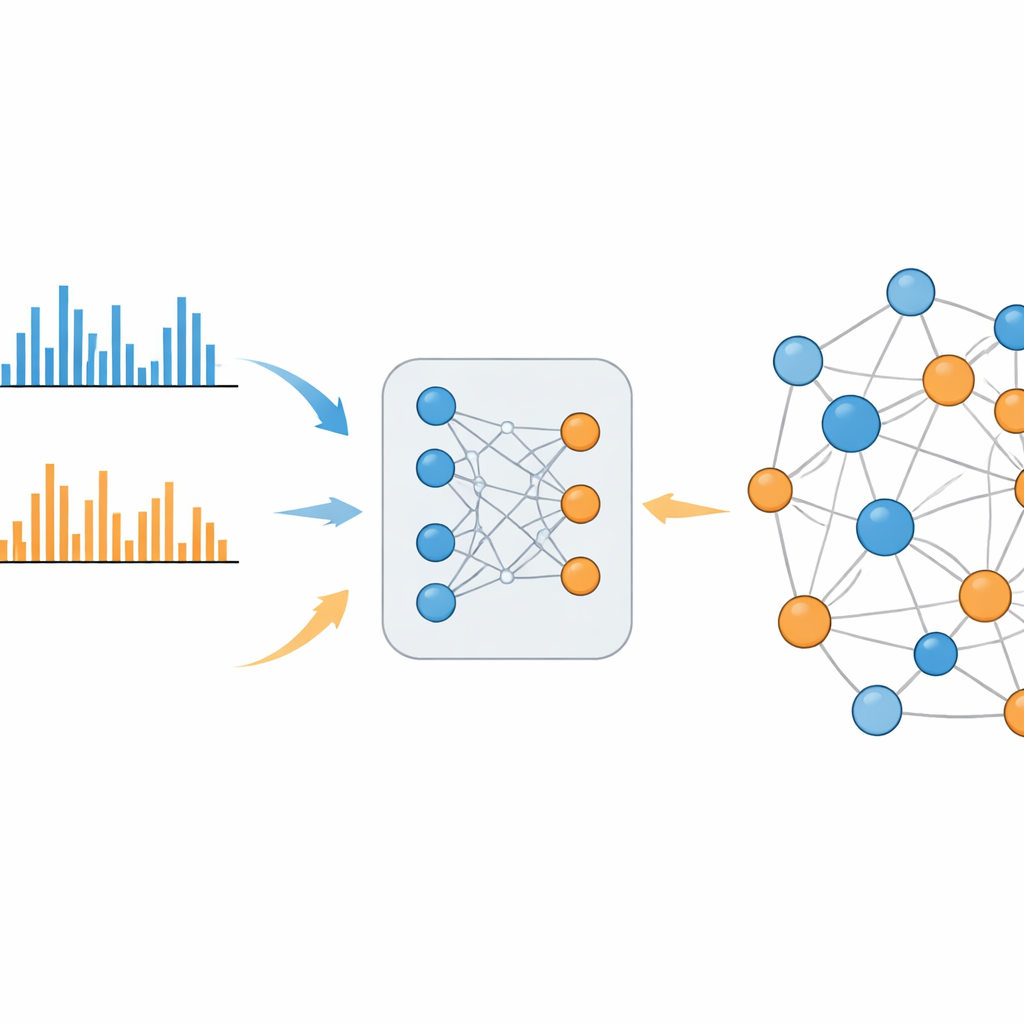
A learning system that bridges the gap
MS2DeepScore 2.0 tackles this divide by learning chemical similarity directly from large libraries of known spectra. The model is built on a twin neural network design that converts each fragmentation pattern into a 500-number fingerprint, called an embedding. During training, the system sees hundreds of thousands of examples from both positive and negative modes, along with how similar the underlying molecules actually are. It adjusts itself so that spectra from related molecules end up with similar embeddings, whether they were measured in the same mode or in opposite modes. The new version goes beyond its predecessor by feeding in extra information, such as the mass of the original molecule and which ionization mode was used, and by using a carefully balanced sampling scheme so that rare but informative chemical relationships are not drowned out by common, uninformative ones.
From scattered signals to unified maps
Once trained, MS2DeepScore 2.0 can estimate how chemically similar any two spectra are, including positive versus negative mode pairs. The authors show that these predictions correlate well with established measures of structural similarity, not only within each mode but also across modes. Using real data from human urine, human blood plasma, and a wild edible plant, they build “molecular networks” in which each spectrum is a node and strong predicted similarity creates a connection. Unlike older approaches, these networks naturally mix positive and negative mode data into single, coherent maps. Expert-curated clusters include, for example, groups of caffeine-related molecules in urine that are linked across ionization modes and match known metabolic pathways.
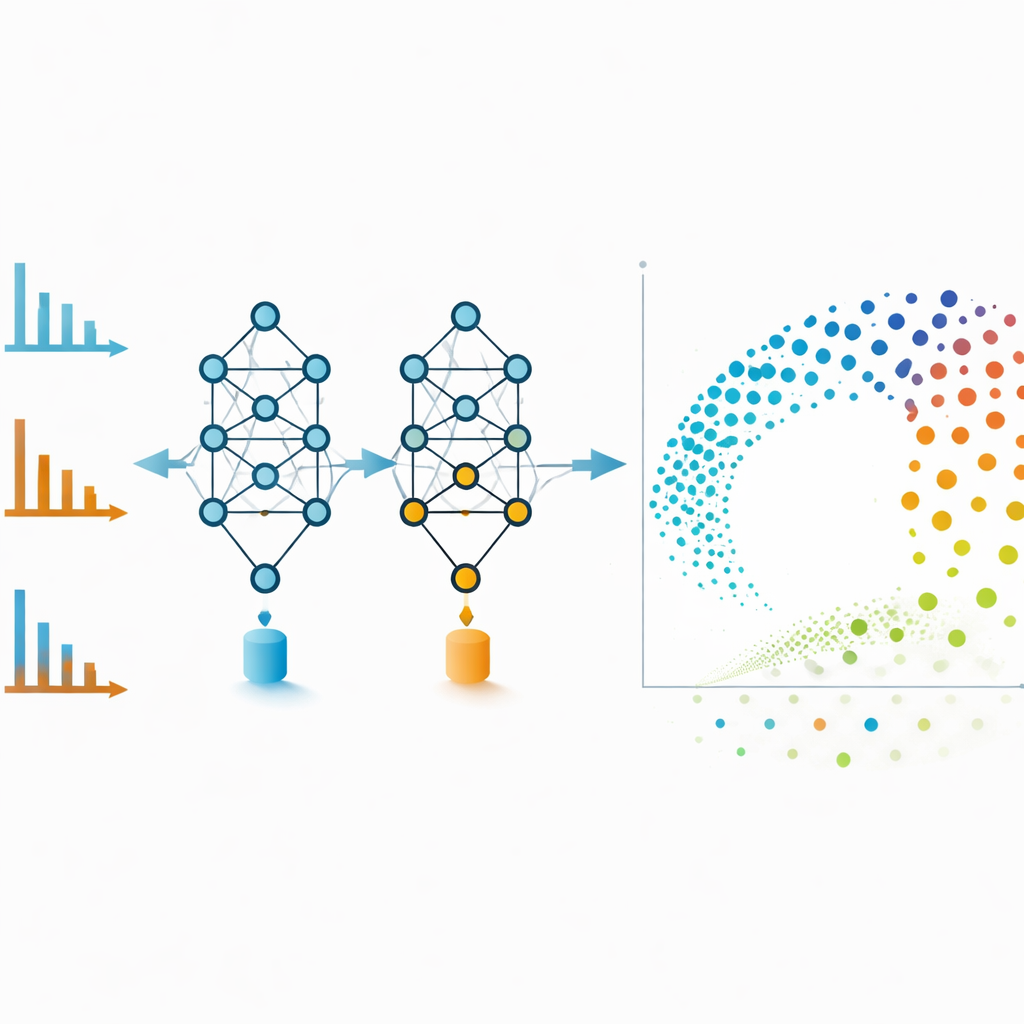
Seeing the chemical landscape at a glance
Molecular networks are powerful but can become tangled if too many weak links are included. To avoid this, the authors use MS2DeepScore’s embeddings directly as coordinates in a two-dimensional layout created with a technique called UMAP. Each dot in this map represents one spectrum, and nearby dots correspond to molecules that the model considers chemically similar. Positive and negative mode spectra of the same compound, which look very different by eye, often end up side by side in this embedding space. The team also trains an additional model that inspects each embedding and estimates how reliable it is, flagging spectra that are noisy, incomplete, or unlike anything seen during training. Removing these low-quality points improves overall accuracy and makes the visualizations more trustworthy.
Bringing advanced tools to everyday labs
To ensure that this technology is usable beyond programming experts, the authors have integrated MS2DeepScore 2.0 into popular, freely available mass spectrometry software. With this integration, researchers can detect features, build molecular networks that ignore ionization-mode boundaries, and explore the resulting chemical space through interactive dashboards. The code, trained models, and example datasets are openly shared, and the system can be retrained or fine-tuned for specialized chemical classes.
What this means for future discoveries
For non-specialists, the key message is that MS2DeepScore 2.0 helps turn fragmented and mode-dependent measurements into a single, more understandable picture of the molecules present in a sample. By reliably linking signals that used to live in separate analytical worlds, the method lets scientists exploit much larger reference libraries, compare samples more completely, and focus their attention on meaningful clusters of related compounds. This cross-connection of data is expected to speed up the identification of biomarkers, nutrients, natural products, and pollutants, ultimately deepening our understanding of how chemistry shapes health and the environment.
Citation: de Jonge, N.F., Chekmeneva, E., Schmid, R. et al. Cross ionization mode chemical similarity prediction between tandem mass spectra in metabolomics. Nat Commun 17, 2483 (2026). https://doi.org/10.1038/s41467-026-69083-y
Keywords: metabolomics, mass spectrometry, machine learning, molecular networking, chemical similarity