Clear Sky Science · en
Supercharging-enhanced nDIA-MS enables global profiling of drug-induced proteome solubility shifts
Why protein behavior matters beyond simple counts
Medicines can change not only how much of a protein a cell makes, but also where that protein goes and whether it floats freely or clumps onto cellular structures. Those shifts in “where” and “how” proteins exist inside cells are closely tied to diseases such as cancer and neurodegeneration, yet they are hard to measure at scale. This study introduces a fast, powerful way to scan thousands of human proteins at once to see how their solubility – whether they are in a free or more stuck state – changes when cells are exposed to two widely used experimental drugs.
A faster, sharper way to look at proteins
The authors build on a core technology of modern biology: mass spectrometry, a technique that weighs and counts protein fragments. Standard methods already excel at telling us how much of each protein is present, but they struggle to reveal changes in protein state, such as moving onto DNA or into dense assemblies. Here, the researchers refine a version of mass spectrometry called narrow-window data-independent acquisition (nDIA-MS). By adding small chemical helpers, called supercharging reagents, to the liquid that carries peptides through the instrument, they boost the electrical charges on these fragments and greatly strengthen the signal.
After testing two common supercharging additives, dimethyl sulfoxide (DMSO) and m-nitrobenzyl alcohol (mNBA), they find that 3% DMSO gives the largest overall signal increase and the highest number of proteins detected, while mNBA is better at increasing the number and charge of individual peptide fragments. With the optimized setup, the team can identify about 9,600 human proteins from just one microgram of a standard cell digest in a 15‑minute run – a remarkable speed and depth combination for routine experiments. This performance sets the stage for using nDIA-MS not only to count proteins, but to probe how their physical state responds to stress.
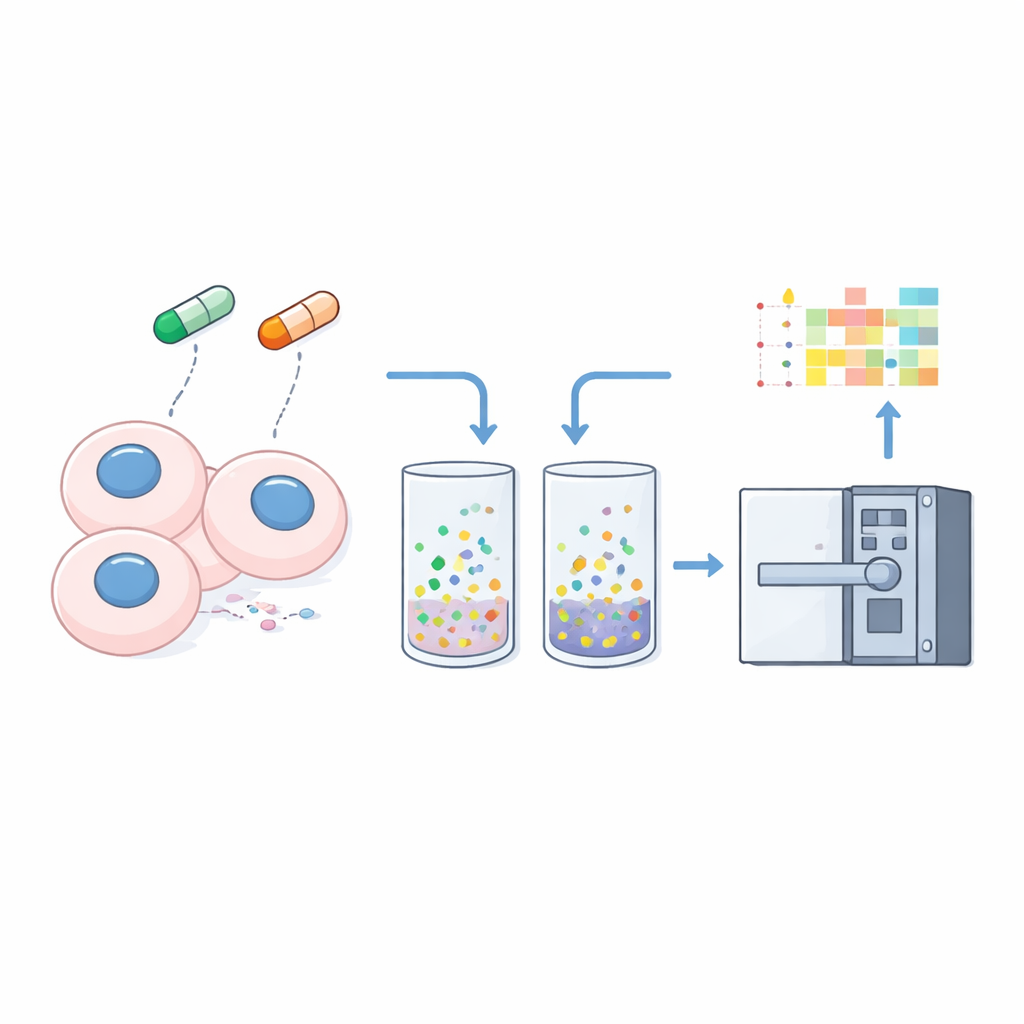
Turning solubility into a readout of cellular change
To transform solubility into a measurable signal, the researchers gently pre-treat cells with a mild detergent that extracts freely soluble proteins, leaving behind material that is tightly bound to DNA, membranes, or other structures. They analyze both the whole-cell lysate and this “insoluble” pellet from three cancer cell lines using the optimized nDIA-MS method. By comparing the abundance of each protein in the pellet relative to the whole-cell sample, they calculate a simple “insolubility ratio” – higher values suggest stronger association with cellular structures, lower values suggest a more soluble, free state.
The team then exposes cells for just one hour to two drugs: MG132, a proteasome blocker that prevents cells from degrading unwanted proteins, and ML‑792, an inhibitor of SUMO activation that interferes with a key protein-tagging system. This short exposure is deliberate; it aims to capture rapid, early shifts in protein behavior before gene expression changes have fully unfolded. Even in this brief window, they can quantify solubility and abundance changes for 8,694 proteins and confirm that their fractionation cleanly separates classic soluble proteins, like tubulin, from chromatin-bound proteins, like histones.
Drugs that reshape the cell’s protein landscape
MG132 and ML‑792 produce widespread but distinct remodeling of protein solubility. MG132 makes over a thousand proteins more insoluble and more than six hundred more soluble across the three cell lines. Many affected proteins sit at the crossroads of protein quality control, DNA damage response, gene regulation, and autophagy – the cell’s self‑cleaning system. For example, components of the proteasome activator complex and adaptors that help tag faulty proteins become less soluble, hinting that when degradation is blocked, key quality‑control factors themselves get trapped on structures such as chromatin or cellular membranes. Autophagy proteins and damage-response players, including the stress regulator HSF1, also shift toward more insoluble, condensate-like states, consistent with the formation of nuclear stress bodies.
ML‑792, in contrast, strongly targets proteins involved in transcription and in the SUMO system itself. Hundreds of proteins become either more insoluble or more soluble when SUMO activation is blocked, and core SUMO proteins move into the soluble pool, as expected. The study highlights striking behaviors: subunits of RNA polymerase III become more insoluble, suggesting altered nuclear import or chromatin binding; key repressors associated with nuclear bodies, such as SP100 and DAXX, become more soluble, indicating that SUMO marks help keep them sequestered in these compartments. When both drugs are applied alone or in sequence, some proteins, including NAB2, SMAD2, and RB1, show coordinated or even opposing solubility shifts, revealing a nuanced interplay between the ubiquitin and SUMO tagging systems in controlling protein localization.
What this means for future drug discovery
For non-specialists, the central message is that proteins do not merely turn up or down in amount when cells are stressed or treated with drugs; they also move, condense, and change how tightly they are tied to cellular structures. This work delivers a high-speed, proteome-wide way to watch those shifts happen. By combining a tuned mass-spectrometry workflow with a simple soluble-versus-insoluble comparison, the authors show that common experimental drugs rapidly reorganize the internal protein landscape in ways that standard abundance measurements would miss. The approach opens the door to mapping how candidate medicines reshape protein states throughout the cell, helping researchers uncover unexpected targets, stress pathways, and failure points that could be crucial for understanding both therapeutic action and side effects.
Citation: Xiong, Y., Zhang, H., Tan, L. et al. Supercharging-enhanced nDIA-MS enables global profiling of drug-induced proteome solubility shifts. Nat Commun 17, 2350 (2026). https://doi.org/10.1038/s41467-026-69025-8
Keywords: proteome solubility, mass spectrometry, proteasome inhibitor MG132, SUMOylation inhibitor ML-792, protein state transitions